
Application of sequence-based methods in human microbial ecology
Abstract
Ecologists studying microbial life in the environment have recognized the enormous complexity of microbial diversity for many years, and the development of a variety of culture-independent methods, many of them coupled with high-throughput DNA sequencing, has allowed this diversity to be explored in ever-greater detail. Despite the widespread application of these new techniques to the characterization of uncultivated microbes and microbial communities in the environment, their application to human health and disease has lagged. Because DNA-based techniques for defining uncultured microbes allow not only cataloging of microbial diversity but also insight into microbial functions, investigators are beginning to apply these tools to the microbial communities that abound on and within us, in what has aptly been called “the second Human Genome Project.” In this review we discuss the sequence-based methods for microbial analysis that are currently available and their application to identify novel human pathogens, improve diagnosis of known infectious diseases, and advance understanding of our relationship with microbial communities that normally reside in and on the human body.
Sequence-based methods
It has long been recognized that standard culture methods fail to adequately represent the enormous microbial diversity that exists in nature because of the fastidious growth requirements of many microorganisms. Even when growth conditions are altered to mimic environmental nutrient composition, up to 80% of organisms identified by culture-independent methods fail to grow in culture (Connon and Giovannoni 2002). To avoid reliance on cultivation, many culture-independent methods have been developed to search for novel bacterial species, including pathogens. These methods include screening of expression libraries with immune serum, nucleic acid subtractive methods, and small molecule detection with mass spectroscopy, among others, and these methods have been reviewed elsewhere (Relman 2002). This review will focus primarily on sequence-based methods because of their general applicability and the continued expansion of high-throughput, low-cost sequencing capacity.
The cornerstone of culture-independent identification of bacterial and archaeal species is sequence analysis of ribosomal RNA genes that are sufficiently well conserved across species that they can be amplified using PCR primers based on highly conserved sequences, yet are sufficiently diverse to differentiate bacterial or archaeal species. Carl Woese, in a series of seminal studies, initially used small subunit (16S) rRNA gene sequences for construction of phylogenetic trees of cultivated organisms (Woese and Fox 1977; Woese 1982; Gupta et al. 1983; Woese and Olsen 1986), but this method was subsequently applied to libraries of rRNA genes PCR-amplified from environmental DNA samples without cultivation (Stahl et al. 1984, 1985; Giovannoni et al. 1990; Ward et al. 1990; Schmidt et al. 1991). A striking collective result from the application of this technique to numerous environmental samples was the realization that cultivated organisms represent a tiny fraction of species present in most environmental samples. Indeed only half of 52 currently recognized bacterial phyla contain cultivated members (Hugenholtz et al. 1998; Rappe and Giovannoni 2003). To maximize the utility of 16S rRNA gene analysis for species determination, it is now routine to amplify almost the entire 16S rRNA gene, which spans ∼1500 bp and is thus readily sequenced in its entirety through bidirectional sequencing of cloned 16S amplicons (Hugenholtz 2002). After sequencing, 16S sequences are clustered into groups and a threshold of sequence similarity is established (usually 98 or 99%) to distinguish species (Fig. 1).
Although PCR amplification of 16S sequences has been of enormous value, there are caveats to this approach. One is that organisms that carry sequence differences within the highly conserved regions used for primer design may amplify less efficiently or not amplify at all. For example, the 16S rRNA gene sequence of the Nanoarchaeota is so divergent that PCR with the “universal” primers failed to detect this species even from cultured organisms (Huber et al. 2002). Second, PCR conditions, such as annealing temperature or extension time, may allow formation of chimeras or produce amplification bias that skews the representation of each species in cloned libraries (Wang and Wang 1997; Kroes et al. 1999; Ishii and Fukui 2001). Some of these errors may be recognized and corrected by hybridization-based methods such as in situ hybridization with species- or strain-specific 16S oligonucleotides applied to the original (or similar) sample.
A relative drawback of 16S rRNA gene sequencing is the need for significant sequencing capacity that, except in high-throughput sequencing centers, may be relatively slow compared with hybridization-based methods. As an alternative, several strategies employing 16S rRNA gene microarrays have been presented and offer some advantage in speed compared with sequencing when analysis of many samples is required (Guschin et al. 1997; Rudi et al. 2000; Small et al. 2001; Loy et al. 2002, 2005). For the most part these studies employed oligonucleotide probes that were designed for detection of specific organisms, such as sulfate-reducing bacteria or beta-proteobacteria, and have offered acceptable sensitivity. Application to highly complex environmental samples has been limited by sensitivity and difficulties in differentiating related species, but it seems reasonable to expect further improvement in this technology and eventual application to clinical materials from humans.
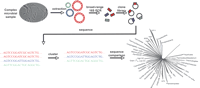
Broad-range PCR amplification and sequencing of microbial 16S rRNA genes. Genomic DNA extracted from a microbial community is used as a template for 16S rRNA PCR with “universal” primers specific for Archaea and Bacteria. The PCR products, which are about 1.5 kb in length, are cloned into a standard vector and both ends are sequenced. The aligned sequences are first clustered into groups, and the representatives from each cluster are compared with 16S rRNA gene databases for phylogenetic classification.
A final drawback to 16S rRNA gene sequencing is the absence of functional genomic information obtained. Recently genomic libraries have been created directly from DNA extracted from environmental samples and subjected to functional screens or to shotgun sequencing with the goal of assembly for the most abundant genome(s) present (Handelsman 2004). These “metagenomic” methods circumvent the needs for cultivation or PCR amplification. Recent metagenomic studies of an acid mine biofilm and the Sargasso Sea yielded significant insight into species diversity and ecology of uncultivated microbial communities (Tyson et al. 2004; Venter et al. 2004; Tringe et al. 2005). While not yet applied to clinical environments, metagenomic methods have the potential to provide functional characterization of complex, human-associated microbial communities.
Identification of novel human pathogens
An exciting application of culture-independent methods is the identification of uncultivated organisms that cause human disease. Because DNA can be extracted from any potentially infected material and used as a substrate for 16S rRNA gene amplification, Fredericks and Relman (1996) predicted that a rash of claims for disease causation by new pathogens would follow application of this method to human tissues and laid out a strategy for proving disease causation for organisms that might be difficult or impossible to grow. Reiman and Falkow (2001) later amplified these criteria. Remarkably, this has not come to pass and only a few new pathogens have been identified through 16S rRNA gene amplification. This may be because present methods for cultivation are sufficient for the vast majority of pathogens capable of growth on human tissues, or it may be that we have yet to apply culture-independent methods to many clinical conditions that have an infectious component. Whatever the reason, these new infectious agents are notable and worthy of review here.
The first novel pathogen to be identified by sequence-based methods was Rochalimaea henselae, the organism responsible for bacillary angiomatosis (BA). The hallmark of BA is abnormal proliferation of small blood vessels in the skin and visceral organs of immunocompromised patients. Although bacteria had been found in tissue sections by Warthin-Starry staining, they could not be cultured because of their fastidious growth requirements (Perkocha et al. 1990). In 1990, Relman et al. amplified a partial 16S rRNA gene sequence from tissue samples obtained from bacillary angiomatosis patients, but not from normal tissues, using broad-range PCR (Relman et al. 1990). Analysis of this 16S sequence suggested a novel species most closely related to Rochalimaea spp. Further evidence for causation was provided by the isolation of a slow-growing, Rochalimaea-like bacillus from a single BA patient in an independent study (Slater et al. 1990). Two years later, genotypic analysis of the complete 16S rRNA gene and other genomic loci further confirmed the novelty of the isolated BA agent, Rochalimaea henselae (later moved into the genus Bartonella) (Regnery et al. 1992).
The same strategy was soon applied to other potentially infectious diseases and led to the identification of Ehrlichia chaffeensis, a new species associated with tick bites that causes a febrile illness. Ehrlichiosis is clinically similar to Rocky Mountain spotted fever, another tick-borne disease caused by the intracellular parasite, Rickettsia rickettsii. Although testing of the index patient's serum for antibodies against R. rickettsii was negative, patient serum contained antibodies reactive to E. canis, a well-described canine pathogen (Maeda et al. 1987). This suggested that E. canis or a related species was responsible for disease, and 16S rRNA gene amplification from infected macrophages led to identification of E. chaffeensis (Anderson et al. 1991; Dawson et al. 1991). Causation is supported by concordance of E. chaffeensis 16S rRNA and serologic findings in patients with fever, leukocyte inclusions, and history of a tick bite, as well as a salutary clinical response to appropriate antibiotics accompanied by loss of E. chaffeensis 16S rRNA gene from leukocytes.
A third example of success with 16S rRNA gene amplification is Whipple's disease. Whipple's disease is a rare disease first described in 1907 in a missionary who died of an illness marked by chronic joint pain, weight loss, and severe abdominal pain. In the report of this patient, “rod-like bacilli in a small node” were noted (Whipple 1907). Eighty-four years passed before the identification of the etiologic agent, despite its consistent observation in affected tissues (Chears Jr. and Ashworth 1961; Yardley and Hendrix 1961) and patients' improvement with antibiotic treatment (Trier et al. 1965). In 1991, a partial 16S rRNA gene sequence was amplified from a small-bowel biopsy specimen taken from a patient with Whipple's disease (Wilson et al. 1991), and the complete 16S rRNA gene sequence was determined a year later, revealing it to be an actinomycete not closely related to any known genus. It was therefore given a new genus and species name, Tropheryma whipplei, based on the unusual features of the disease and the distinct morphological characteristics of the bacillus (Relman et al. 1992). It is worth noting that T. whipplei was particularly recalcitrant to cultivation (Raoult et al. 2000). The complete genome sequence of T. whipplei predicted deficiencies in amino acid synthesis (Bentley et al. 2003; Raoult et al. 2003), and with this information, Renesto et al. (2003) successfully designed a complete medium that allowed cell-free cultivation of T. whipplei. This was the first demonstration that genomic information could guide rational design of media for axenic cultivation of fastidious bacteria.
Whereas most human tissues are normally devoid of cultivable microorganisms, many epithelial-lined cavities of the human body in contiguity with the environment harbor microbial communities, the complexities of which are just beginning to be understood. These include the skin, mouth, ear, gastrointestinal tract, and vagina. Identifying pathogens within this complex bacterial background is more difficult than identifying them in normally sterile compartments. One of the most successful examples of this involves the study of dental plaque. Because of their known role in dental caries and periodontal disease, human oral flora have been studied intensively through both culture-dependent and culture-independent techniques. About 500 bacterial species have been found in the human oral cavity (Thoden van Velzen et al. 1984; Meyer and Fives-Taylor 1998; Paster et al. 2001) and 40%–60% of these species are uncultivated “phylotypes” (Kroes et al. 1999; Paster et al. 2001). Studies using conventional culture methods have established that early colonizing streptococci play a key role in initiating the formation of dental caries (Kolenbrander et al. 1990; Whittaker et al. 1996), while Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis and Treponema denticola contribute to the development of periodontal disease (Meyer and Fives-Taylor 1998). Recent 16S rDNA sequence analysis uncovered that in addition to these known pathogens, several additional organisms, including organisms assigned to uncultivated phyla OP11 and TM7, are strongly associated with periodontitis or acute necrotizing ulcerative gingivitis in humans (Choi et al. 1994; Dewhirst et al. 2001; Paster et al. 2001; Brinig et al. 2003; Hutter et al. 2003; Ouverney et al. 2003).
Recently, methanogenic Archaea have also been linked to periodontal disease based on 16S rRNA sequencing and FISH analysis (Kulik et al. 2001; Lepp et al. 2004), providing the first example of archaeal disease association in humans. In the latter study, the relative abundance of methanogenic Archaea was associated with disease severity and decreased with effective treatment (Lepp et al. 2004). However, Archaea were not uniformly found in the subgingival space of severely affected individuals, leaving open the question of whether the Archaea are playing a causative role in this polymicrobial disease. Interestingly, the abundance of Archaea and T. denticola were inversely correlated, suggesting that they might compete for the same niche in the community. It was hypothesized that both organisms serve as “hydrogen sinks” in the highly reduced environment of the subgingival space, allowing acid-producing members of the community to grow to higher density than they might in the absence of Archaea or Treponemes. While additional genomic and metabolic studies will be required to fully understand the role of methanogenic Archaea in periodontal disease, this example clearly illustrates how culture-independent methods may do more than increase our appreciation of the number of known pathogens. These methods also stand to teach us a great deal about the mechanisms of disease, especially in circumstances where the paradigm of a single causative agent seems not to hold. Other clinical circumstances that may benefit from similar analyses include sinusitis, ventilator-associated pneumonia, small-bowel overgrowth syndromes, inflammatory bowel disease, and bacterial vaginosis.
Novel viral pathogens
Although it is unclear how many new bacterial pathogens remain to be identified, it seems likely that a larger number of viral pathogens have thus far escaped detection. This is because growth conditions are far harder to determine and nucleic acid techniques based on sequence conservation are not available. Nonetheless there are several examples of successful application of culture-independent methods to the identification of novel viral pathogens. These include the use of DNA subtraction techniques to identify the Kaposi's sarcoma virus (Chang et al. 1994), expression screening of a cDNA library with patient serum to identify the hepatitis-C agent (Choo et al. 1989), and more recently hybridization-based screening to identify the SARS virus as a coronavirus (Ksiazek et al. 2003). The last example was particularly notable because novel viral sequences were recovered from individual elements of the array to which they hybridized and because the analysis was accomplished with remarkable speed.
The diversity of viral genomes clearly complicates the search for novel pathogens and development of new strategies may be required. One such strategy might employ large-scale sequencing of appropriately extracted clinical materials on a massively parallel, single-molecule sequencing apparatus. Sequencing machines capable of producing >200,000 short sequencing reads in a single run, from small amounts of DNA without prior cloning, are now commercially available (Andries et al. 2005). Such machines produce sequence at ∼20% the cost of Sanger sequencing on capillary sequencers and the short reads produced by single-molecule techniques provide an efficient way of producing molecular tags for microbial species. Infected tissues or body fluids could be extracted, reverse transcribed if needed (for RNA viruses), and sequenced along with contaminating host or bacterial DNA. The resulting sequence reads would then be searched for homology to known viral (and bacterial) sequences. Viral sequences found could then be linked by subsequent PCR or used in hybridization-based strategies to recover complete viral genomes.
Sequence-based methods for rapid diagnosis of bacterial infections
Sequence-based methods have also found application to the rapid identification of human pathogens that can be cultured. For fastidious or slow-growing organisms the advantage is obvious, but there may be significant value in their application to more common infectious agents because standard culture techniques require 24–48 h for growth and identification of most bacterial species. Clinical practice has long favored the use of antibiotics to cover the organisms most likely to be present when infection is suspected, but this practice has contributed greatly to the spread of antibiotic resistance (Neu 1992; Cohen 2000). Hence, the use of culture-independent methods for rapid diagnosis has the potential to provide more specific antibiotic therapy from the outset or to withhold it altogether if there is no infection present. Because of the continuing need for analysis of antibiotic sensitivity, culture is still required for every serious infection, but one can envision a day when resistance is sufficiently well understood that this might also be rapidly predicted from nucleic acid-based assays.
Both hybridization and PCR-based strategies have been used for rapid diagnosis. 16S rRNA gene amplification can be carried out using universal primers followed by detection with group-specific fluorescent probes or a second group-specific PCR. When applied to tissues or fluids that are normally devoid of cultivable organisms, including blood, urine, cerebrospinal fluid, wounds, and indwelling intravascular catheters, these assays have generally been able to identify more than 95% of infected samples with false–positive rates of ∼10%. (Qin and Urdahl 2001; Nikkari et al. 2002; Domann et al. 2003; Moumile et al. 2004; Schuurman et al. 2004). While these studies demonstrate acceptable performance, to our knowledge, no one has yet fulfilled the promise of these methods to guide therapy prospectively or to reduce the administration of broad-spectrum antibiotics to potentially infected patients while awaiting culture results.
Understanding complex microbial communities in human ecology
It is increasingly clear that humans have a symbiotic relationship with several microbial consortia living on and within us, but understanding the details of these relationships is a major challenge. One important application of culture-independent methods has been preliminary evaluation of these communities. In the discussion below we will focus on the gut microbial consortia because it clearly illustrates the complexity of the problem in a context where recent progress has been made.
It has long been appreciated that the human gastrointestinal tract is colonized by a complex microbial community whose numbers are believed to far exceed the total number of cells in the human body. Although this community has been studied in detail by relatively efficient culture techniques, culture-independent studies suggest that 40%–80% of the total microscopic counts are uncultivated species (Langendijk et al. 1995; Suau et al. 1999). Several studies have used 16S rRNA gene amplification to uncover numerous novel species (Wilson and Blitchington 1996; Suau et al. 1999). Quantitative assays of human fecal samples collected from several individuals have shown that each person has a unique collection of bacteria, and an individual's dominant flora is relatively stable over time (Franks et al. 1998; Zoetendal et al. 1998). These findings were recently confirmed with a large-scale 16S survey, which also found 62% of bacterial phylotypes in the gut were novel and 80% represented uncultivated species, mostly from the Firmicutes and Bacteroidetes phyla (Eckburg et al. 2005).
The finding of greater similarity between gut microbes of monozygotic twins living apart than genetically unrelated individuals living together suggests that host genetics may significantly affect the composition of gut microbial flora (Zoetendal et al. 2001). A recent study demonstrated expansion of segmented filamentous bacteria in the gut of IgA-deficient mice compared with wild-type litter mates, confirming that host genotype molds the gut microbial consortium and suggesting that the host immune system is responsible for regulating normal flora in addition to protecting against pathogens (Suzuki et al. 2004).
Studies in mice, rats, and fish raised in sterile environments, and therefore lacking this community in the gut, have demonstrated the importance of this community for normal gut structure and function, nutrient absorption, fat deposition, and development of normal immunity (Falk et al. 1998; Guarner and Malagelada 2003; Backhed et al. 2004, 2005; Rawls et al. 2004). For example, in the recent study by Rawls et al. (2004), gene expression patterns were compared between germ-free and normal gut in both mice and zebrafish, and 59 genes were identified with concordant changes in the two species. One gene that was potently suppressed by the presence of gut microbiota in both mice and fish is Fiaf, an inhibitor of lipoprotein-lipase, a gene important for fat accumulation. Reconstitution of gut microbiota increased body fat by more than 50% in germ-free mice. However, inactivation of Fiaf by gene targeting eliminated the normal microbiota-induced deposition of fat, demonstrating an important role for this protein in microbe-induced fat deposition. The mechanism by which gut microbial flora suppress Fiaf expression is unknown (Backhed et al. 2004).
Thus, it appears that the human gut exhibits a true symbiosis in which the microbial community enjoys a stable, nutrient-rich environment with a limited host immune response, and in turn, the microbes selected to exist in this environment facilitate normal gut function. Unfortunately, although 16S rRNA gene surveys have successfully defined microbial diversity in the gut, these studies provide little functional information to help us understand specific microbial functions relevant to human health. Dissecting these functions is complicated for several reasons. First, microbial diversity within and between individuals makes complete description of the community difficult. A complete description is important because minor species may provide important functions. Second, the number of sequenced genomes from the microbial community relative to the total number of species is relatively limited, and third, genomic tools for predicting novel functions of complex microbial communities have not existed.
An alternative strategy for understanding potential functions of microbial communities that circumvents these problems was recently reported by Tringe et al. (2005) for analysis of several environmental microbial communities (and Fig. 2). This analysis represents the microbial community not from the viewpoint of the individual organisms present but rather from the collective gene content of the constituent organisms. For this analysis, small-insert libraries were created for soil and several deep-ocean whale carcasses and approximately 25–100 Mb of sequence was generated from each library. No attempt was made to assemble the sequencing reads (each ∼700 bp in length). Instead, the reads (referred to as “environmental gene tags” or EGTs) were individually annotated and when possible a putative gene function was assigned to each read based on its best BLAST hit. EGTs were then binned by function and the prevalence of specific functions was compared between four environments: soil, whale falls, and two previously reported environmental samples, a biofilm obtained from acid mine runoff (Tyson et al. 2004) and seawater collected from the Sargasso Sea (Venter et al. 2004). Trees constructed from these EGTs showed that samples obtained from similar environments clustered together. Furthermore, analysis of EGTs overrepresented in specific environments indicates they perform functions important for survival in that environment (e.g., sodium transporters in seawater). It is easy to imagine how such an analysis could be used to understand the function of microbial community in the gut and other human environments. In a fashion analogous to expression profiling of mammalian tissues, EGT fingerprints of microbial communities from diseased and normal individuals might be compared, or those from single individuals subjected to different diets.
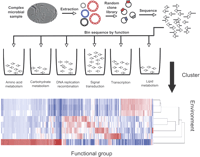
Environmental genomic tags for functional analysis of complex microbial communities. Genomic DNA is extracted from the community, sheared, and cloned into a standard sequencing vector. Clones are sequenced and function assigned to individual reads based on highly significant BLAST hits. These reads are then binned by function, and the relative abundance of reads in each functional category can be clustered and displayed graphically so that multiple environments can be compared. The clustering algorithm also computes and displays branch lengths that represent relatedness of individual samples.
With expanding sequence capacity and new technologies like EGT fingerprints, the value of studying microbial communities in genetically tractable model organisms seems clear. Very recently, Ley et al. (2005) reported the finding of the cecal microbiota composition change in genetically obese (ob/ob) mice using 16S rRNA sequence analysis. The obese mice, which carry a homozygous mutation in the leptin gene (ob/ob) locus, displayed a statistically significant reduction in Bacteriodetes when compared with their lean (ob/+ or +/+) siblings. This result suggested that obesity may affect the diversity of the gut microbiota at division level in nearly isogenic mice. However, the mechanism for such changes remains unknown. Comparing gene content of gut microbes in ob/ob mice and lean siblings through a metagenomic approach might provide novel insights in understanding how changes of community structure at division level may affect energy homeostasis. While the study of Tringe et al. (2005) compared quite different microbial communities, we expect that the number of EGTs altered in congenic mice, raised together and eating the same diet but differing in the presence or absence of a single gene, should be much more limited and could be readily related to phenotypic changes in the host.
Conclusions and future directions
Culture-independent methods, while largely developed for analysis of environmental microbes, have found broad utility in human ecology and have led to an appreciation for the diversity of microbial communities inhabiting normal humans. Quantitative PCR methods show particular promise for providing rapid identification of human pathogens that may allow clinicians to narrow and limit antibiotic use, which could in turn limit the spread of antibiotic resistance. As DNA-sequencing capacity grows and costs fall, sequence-based methods of analysis will be expanded to provide snapshots of the microbes present in many body fluids and tissues and the functions encoded in their genes. Specifically, we predict that expansion of EGT-based methods to complex microbial communities in the mouth, gut, skin, and vagina may lead to an understanding of their role in human health and disease.
Acknowledgments
This work was performed under the auspices of the U.S. Department of Energy's Office of Science, Biological, and Environmental Research Program and by the University of California, Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48, Lawrence Berkeley National Laboratory under contract No. DE-AC03–76SF00098, and Los Alamos National Laboratory under contract No. W-7405-ENG-36 and was supported by NIH-NHLBI THL007279F. We thank Phil Hugenholtz, Susannah Tringe, Tanja Woyke, and members of the Rubin laboratory for their critical reading of the manuscript.
Footnotes
-
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.3676406.
-
↵1 Corresponding author. E-mail jbristow{at}LBL.gov; fax (925) 296-5752.
-
- Accepted October 23, 2005.
- Received January 10, 2005.
- Cold Spring Harbor Laboratory Press








