
The mouse genome
Abstract
The house mouse has been used as a privileged model organism since the early days of genetics, and the numerous experiments made with this small mammal have regularly contributed to enrich our knowledge of mammalian biology and pathology, ranging from embryonic development to metabolic disease, histocompatibility, immunology, behavior, and cancer. Over the past two decades, a number of large-scale integrated and concerted projects have been undertaken that will probably open a new era in the genetics of the species. The sequencing of the genome, which will allow researchers to make comparisons with other mammals and identify regions conserved by evolution, is probably the most important project, but many other initiatives, such as the massive production of point or chromosomal mutations associated with comprehensive and standardized phenotyping of the mutant phenotypes, will help annotation of the ∼25,000 genes packed in the mouse genome. In the same way, and as another consequence of the sequencing, the discovery of many single nucleotide polymorphisms and the development of new tools and resources, like the Collaborative Cross, will contribute to the development of modern quantitative genetics. It is clear that mouse genetics has changed dramatically over the last 10-15 years and its future looks promising.
Because of its many advantages as an animal model, geneticists have used the house mouse since the early days of genetics. Historical records indicate that Mendel himself bred and crossed mice, segregating for coat color mutations, until he was requested by the ecclesiastical hierarchy to stop experimenting with animals and to resume working with garden peas (Paigen 2003). In 1902, shortly after Mendel's laws were rediscovered, Lucien Cuénot used mice to demonstrate that the laws in question applied to mammals as they did to plants (Cuénot 1902). Since these initial observations, and if we exclude interruptions in progress due to the world wars, it is not an exaggeration to say that the advances in mouse genetics have been growing exponentially, even booming over the last fifteen years. In this anniversary review, I will cover the most important achievements that occurred in the period spanning 1990 to 2005 and will discuss the anticipated developments in the years to come.
Before we start reviewing these achievements, it is important to note that the context in which mouse genetics has been evolving during the period covered by this review can in no way be compared with the period before it. For the majority of the 20th century, the community of mouse geneticists operated like a club of friends, with occasional meetings and the exchanging of ideas and animals (mostly mutant strains) in a very informal way. During this period, research projects were run on a small scale and were carried out independently. With the advent of molecular techniques for mouse genetics, and in particular after the development of in vitro transgenic techniques that made it possible to manipulate the genome almost at will, the situation changed dramatically. The number of scientists working with mice increased abruptly, probably because the community realized that engineering the mouse genome was perhaps the most efficient way to study gene function and to generate animal models for human pathologies. During the same period, several large integrated and concerted projects, sometimes on an international scale, were undertaken, and these resulted in an enormous increase in our knowledge of the species. These projects included the sequencing of the mouse genome, the production of thousands of new mutations with chemicals or by gene trapping, the accurate and systematic phenotyping of many inbred strains, the development of tools aimed at a better analysis of complex traits, as well as a few other projects, all of which have opened a new era in mouse genetics. There is no reason to believe that, after such a boom, the situation will enter into a recession. Another radical change was that an enormous amount of new information was made available to the community immediately after being gathered through a network of databases that were easily accessible on the Web and free of charge (see Table 1 for a listing of some of these databases). Indeed, the last fifteen years have been crucial for the development of mouse genetics, and they have certainly paved the way for a few decades to come.
Some useful Web sites
Sequencing of the mouse genome and its consequences
The recent publication of the nearly complete mouse genome sequence (Waterston et al. 2002) can be regarded as a major event for two main reasons. First, the availability of this sequence has provided direct access to the blueprint of a living creature that is relatively close to our own species. This has allowed the identification of similarities and differences between humans and mice, from which it is possible to gather information about genome evolution and gene function at the molecular level. Second, easy access to the sequence of the mouse genome assists scientists in designing more efficient genetic alterations in embryonic stem (ES) cells. In addition to these two points, which were expected and even listed among the arguments in favor of sequencing, the accessibility of a nearly complete and reliable sequence of the mouse genome has had several other important consequences for geneticists by providing them with an enormous number of new polymorphisms.
The first drafts of the mouse sequence, released shortly after the turn of this century, were of excellent quality with error rates lower than 10-4. However, they contained several gaps that were due to inherent limitations in the sequencing protocol (whole-genome shotgun) (Waterston et al. 2002) and to technical difficulties encountered for certain genomic segments, such as duplications or highly repeated regions (Bailey et al. 2004). Many of these imperfections are now being corrected, and the latest assembly released by the Mouse Genome Sequencing Consortium (NCBI build 34) has a length of 2.6 Gb, of which about 1.9 Gb (73%) is finished with less than one sequencing error per 10-5 base pairs. A few chromosomes (Chr 2, 4, 11, and X) are entirely sequenced, allowing comparisons with homologous regions of the human genome to be performed at a very high resolution. Such comparisons, revealing similarities and differences, potentially are a rich source of information. Similarities, for example, allow us to detect regions that are under selective pressure (genomicists sometimes say “purifying selection”) and have remained unchanged or nearly so for several millions of years because they are genetically important and, accordingly, have resisted random drift. Differences at the sequence level may be even more interesting a priori, because they may contain keys explaining how speciation proceeds.
Mouse and human genomes are very similar
When comparing the mouse genomic sequence with that of human, the overall impression is one of similarity (Pennacchio 2003). NCBI build 34 indicates that the mouse sequence is about 14% shorter than the human sequence, but a number of segmental duplications, repeats, and nonalignments are still being analyzed, which, when completed, may make the difference less noticeable (Cheung et al. 2003; Fitzgerald and Bateman 2004). In the same way, the genome that has been chosen for sequencing is the one of the C57BL/6 inbred strain, which is the most widely used strain, but it would not be surprising to discover in the end that different inbred strains in fact have a slightly different number of genes. Sequence comparisons at high resolution, made by matching orthologous segments spanning a few tens of base pairs from each genome and scoring the number of nucleotide mismatches at variable stringency, reveal that, on average, 40% of the mouse sequence can be aligned to the human sequence (Waterston et al. 2002; Schwartz et al. 2003). The stringency of sequence conservation is, however, unevenly distributed (Yap and Patcher 2004). Coding sequences appear to be highly conserved, but the degree of conservation varies depending on the function of the protein. This peculiarity, which is associated with our knowledge of the genetic code, has been exploited by informatics experts to validate their estimations of the coding fractions of the mouse and human genomes. They concluded that the number of exons in both species is almost equal (245,200). They also postulated the existence of a very similar number of protein-coding genes (∼25,000 in the mouse and ∼24,200 in human). This is much less than expected a few years ago, but one can trust that these numbers are close to reality since, after major improvements in the computerized analysis of genome sequences (e.g., after detection of the exonic sequences and validation of the actual coding potential of a sample, by RT-PCR amplification in a panel of RNA libraries), these estimations seem to have stabilized (Guigó et al. 2003; Parra et al. 2003).
Another interesting observation is that about 90% of the mouse and human genomes can be partitioned into regions of conserved synteny, reflecting the structural organization of the chromosome in the common ancestor. In fact, the two genomes share about 350 segments of conserved synteny, whose sizes range from 300 kb to 65 Mb, with a mean of 7 Mb. About 99% of mouse genes have a homolog in the human genome, and for 80% of these genes, the best match in the human genome has, in turn, its best match against the orthologous mouse gene in the conserved syntenic interval. This one-to-one ratio allows us to define a set of genes that are mammal-specific: a basic kit of genes for creating a basic mammal! Of course, the precise delineation of this set of genes requires that comparisons are made with several other mammalian genome sequences as they become available (rat, dog, pig, cattle, macaque, chimpanzee, etc.).
At high stringency, the percentage of conservation between the human and mouse sequences is close to 5%, which indicates that other sequences, in addition to those encoding proteins and representing roughly 1.5% of the genome, are under selective pressure. The function(s) of these conserved noncoding sequences (CNSs) is the subject of intense research at the moment, and there is little doubt that CNSs will keep geneticists busy for another few years. It makes sense to speculate that many of these sequences are essential for controlling the correct spatial and temporal expression of genes (regulatory elements). Other CNSs are known to encode a variety of essential nontranslated RNAs. Some of these RNAs, such as tRNAs (about 350 units), ribosomal RNAs, and micro RNAs, have relatively well-known functions. The functions of many others are totally unknown. Finally, some CNSs probably play an important role in the organization of chromosome structure, which is inherited, and also in the determination of imprinting. The regions with the highest levels of sequence conservation are found in certain domains of genes that encode proteins playing an important role in the patterning of development (e.g., the Hox and Pax series). They are also found in some CNSs whose function is yet unknown. Obviously, a precise inventory of these sequences, as well as knowledge regarding their structure and function, will be critical for making comparisons with other species and for engineering specific alterations in vitro in embryonic stem (ES) cells.
The discovery of all these similarities is not so surprising if we consider the relatively short evolutionary distance between the mouse and human species (75 ± 15 Myr). This is important to know because the similarities can be used for making predictions in one species in the regions where the sequence is incomplete or less reliable in the other. They can also be used for making comparisons with a third species (e.g., rat or chimp) when the sequence is complete and reliable but different in human and mouse. This would be especially helpful for scientists whose aim is to decide whether a gene present in one species and absent in the other results from the addition to or deletion from the ancestral chromosome. Finally, similarities at the sequence level will be important to consider for the analysis of other mammalian genomes whose sequences will remain a draft.
Human and mouse genomes exhibit some interesting differences
Comparisons concerning the number of genes in the two genomes need to be interpreted with care because they do not take into account the existence of, for example, nonprocessed pseudogenes (D'Errico et al. 2004). These genes, which originate from a common ancestor either by gene duplication or, less frequently, by unequal crossing over, are more frequent in the mouse genome than in the human genome, and some are functional while others are not (Cheung et al. 2003; D'Errico et al. 2004). A well-known example involves the gene(s) coding for the hormone Renin (Ren1 and Ren2). Some mouse strains (and humans) have only one copy of the gene (Ren1, coding for rennin, primarily in the kidney and, at a very low level, in the submaxillary glands), while other mouse strains have an extra gene (Ren2, which is closely linked to Ren1 and encodes an isoform of Renin in submaxillary glands only). These two genes originated by tandem duplication of an ancestral gene (Panthier et al. 1984). Ren1 is essential, since its invalidation by knockout has deleterious effects, but Ren2 is not. The two genes, however, have different promoters.
A better example to illustrate the complexity in this matter is the case of the OAS (oligoadenylate synthetase) gene cluster, whose function is important in the innate mechanisms of defense against viral infections (Mashimo et al. 2002; Perelygin et al. 2002). In the human, it is a cluster of three genes on chromosome 12, designated respectively as OAS2, OAS3, and OAS1. Human OAS2 and OAS3 correspond to mouse orthologs Oas2 and Oas3 (Chr 5), and the transcription products of these genes are very similar, with two alternatively spliced isoforms encoded in both mouse Oas2 and human OAS2 and only one transcript from human OAS3 and mouse Oas3. However, the structural organization of OAS1 is very different in the two species, with only one OAS1 gene encoding four different OAS proteins (p42, p44, p46, and p48) in human and no less than eight transcription units in the mouse, which are all orthologous of the OAS1 gene and arranged in tandem with the following order: Oas1e, Oas1c, Oas1b, Oas1f, Oas1h, Oas1g, Oas1a, and Oas1d. For all of these eight genes, a specific interferon-inducible promoter regulates the transcription of a single product, except for Oas1a, where there are two alternatively spliced transcripts (Mashimo et al. 2003). It is likely that a few of these eight genes are nonfunctional pseudogenes that have been generated by duplication of the ancestral OAS1 gene; others, however, look like bona fide genes. We know that a mutation in Oas1b that leads to gene invalidation is correlated with an extreme susceptibility of the affected mice to flavivirus infections (Mashimo et al. 2002; Perelygin et al. 2002). We also know that mice homozygous for a knockout allele of Oas1d (Oas1d-/-) display reduced fertility because of defects in ovarian follicle development (Yan et al. 2005). These two phenotypes are apparently totally unrelated, and the situation is far from being clear.
Situations where the mouse genome harbors genes that are orthologous to human genes but variable in terms of copy number are common. Genes encoding olfactory or taste receptors are such an example; they are arranged in clusters and are at least three times more numerous in mouse than in human. Similar observations have also been made for genes encoding proteins with an immunological function and genes encoding proteins involved in the metabolism of drugs. It has been suggested that these variations resulted from different selective environmental pressures experienced by the ancestors of modern rodents and primates that contributed to “genome shaping” (Godfrey et al. 2004).
If comparisons of the mouse and human genomes allow for detecting such examples of “gene birth,” they also allow for detecting cases of “gene death.” On human chromosome 4, for example, the gene encoding Interleukin 8 (IL8) has no ortholog in the homologous segment of mouse Chr 5 (Fig. 1). IL8 is a chemokine that is secreted by several cell types and is one of the major mediators of the inflammatory response. Surprisingly, although a mouse ortholog of human IL8 is not detectable, Il8rb, which is an ortholog of the gene coding for human IL8 receptor, has been cloned and mapped on mouse Chr 1, and the protein encoded by this Il8rb gene has a very strong similarity to human IL8. For the time being, immunologists have no answer to this puzzling situation. Maybe the mouse uses another gene to achieve the same function or, maybe, the ortholog of human IL8 is transposed elsewhere in the mouse genome. The second explanation seems unlikely because there is no ortholog of human IL8 in the rat genome either. Other examples of gene death have also been reported (Fitzgerald and Bateman 2004).
Comparisons concerning the absolute number of genes in the mouse and human genomes must also be interpreted with care because of important differences in the RNA splicing mechanism. The physical entities that computer scientists detect at the sequence level and label “gene” may encode a different number of proteins in the two species by arranging their set of exons in the transcription products differently. However, what is more important for the biologist is the set of cDNAs (encoding proteins or not) rather than a mere inventory of the genes. To document this point, an international consortium organized and led by the RIKEN Institute in Yokohama, Japan, has undertaken the task of establishing a comprehensive inventory of the cDNA collection encoded in the mouse genome, including functional annotation for each component (Hayashizaki 2003a,b; Carninci et al. 2005). “Deciphering the logic of the transcriptome” is the ultimate goal of the consortium, and the database is known as FANTOM (an acronym for functional annotation of the mouse genome). FANTOM has had three successive releases: FANTOM1, FANTOM2, and FANTOM3. Genome Research published a special issue about FANTOM2 (Vol. 13(6b), June 2003), in which a library gathering a total of 60,770 full-length cDNA clones prepared from 263 mouse tissues that were collected at different stages of development was thoroughly analyzed by the consortium (Okazaki et al. 2002; Kasukawa et al. 2003). This considerable amount of work led to the identification of regions that are potential promoters for 8637 known genes, with an estimation of 63,000 putative transcriptional starting points (Dike et al. 2004). FANTOM3, an even more advanced generation of the database, is now in progress; it aims to identify full-length cDNAs in a total of 103,000 clones (Carninci et al. 2005). All of these databases are regularly updated, are publicly available, and are freely accessible.
Other differences between the mouse and human genomes are worth considering in the frame of this review. Among these, an ensemble of repeated elements known as long interspersed elements (LINES) and short interspersed elements (SINES), as well as the long terminal repeats (LTRs) of retroviruses, are probably the most important or are, at least, the best known (McCarthy and McDonald 2004). These sequences represent about 38% of the total amount of mouse genomic DNA and are active retrotransposons. Some elements move in the genome; they increase in number as they transpose and are occasionally lost in blocks. Genomicists believe that the difference in size between the mouse and human genomes (∼0.3 Gb) might be due, at least in part, to a more efficient mechanism of “transposon cleaning” in the mouse. In fact, transposition of these elements is much more active in the mouse than in the human genome, where transposons have been found to be responsible for about 10% of the spontaneous mutations when they insert inside genes and interfere with the splicing mechanics. The role of these elements as mutagenic agents is quite clear. However, in addition to this role, some scientists in the community believe that transposons may also assume regulatory functions by altering the expression of neighboring genes after transposition (Allen et al. 2004; Medstrand et al. 2005). This point, however, is not yet universally accepted and should be the matter of future investigations. Another amazing peculiarity of these elements is their very heterogeneous distribution in the genomes of both human and mouse, with some homologous regions being totally preserved and others “overcrowded.” This is very puzzling for elements that are, by definition, mobile.

About 90% of the mouse and human genomes can be partitioned into regions of conserved synteny with similar linear arrangements of the genes. Sometimes, however, some segments are either deleted or duplicated. In this case, the gene coding for Interleukin 8 (Il8) is deleted from the mouse genome while the human ortholog (IL8) is still present.
Single nucleotide polymorphisms: An unmatched wealth of polymorphisms
While the genome of the mouse was being sequenced, genomicists discovered that, when homologous regions originating from different laboratory inbred strains were aligned and compared, base-pair mismatches were rather common yet unevenly distributed. Comparing segments of the C57BL/6 inbred strain (the reference strain) with a panel of other inbred strains yielded either high (≥40 mismatches per 10 kb) or low (∼0.5 mismatch per 10 kb) rates of polymorphism with an abrupt delineation between the segments (Wade et al. 2002; Wiltshire et al. 2003). In all strain-to-strain comparisons examined, about one third of the genome appeared to be composed of long regions (≥1 Mb) containing high single nucleotide polymorphism (SNP) rates, while the rest of it exhibited low SNP rates (Fig. 2).
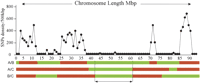
The SNP density between any two inbred strains of mice varies according to the chromosomal region concerned and changes abruptly when passing from one region to the next. This observation is in agreement with historical data on the origin of laboratory inbred strains indicating that they are derived from a small pool of wild ancestors belonging to different subspecies of the genus Mus. Because of this polyphyletic origin, the mouse genome can be regarded as a mosaic of chromosomal segments of various sizes. When the SNP density is low, the segments in question share the same ancestral origin and the few observed SNPs are those resulting from recent mutations. When the SNP density is high, on the contrary, the chromosomal segments have a different origin. When three strains are compared, as on the diagram represented here, one can perfectly observe that three homologous segments have a high SNP density on pairwise comparisons, if all three of them have an independent origin stemming, for example, in three different subspecies of the genus Mus. When a particular region with low SNP density cosegregates with a particular phenotype, the region in question may harbor the genetic determinants for the phenotype in question. (Redrawn with permission from PNAS © 2003, Wiltshire et al. 2003.)
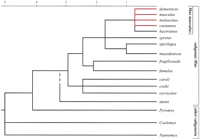
Evolutionary tree of the genus Mus (the time scale is in millions of years, Myrs). The branch leading to the subgenus Mus encompasses all of the species with a basic karyotype of 2N = 40 acrocentric chromosomes. Today's classical laboratory strains are recombinant strains derived (in unequal percentages) from three parental components: Mus musculus domesticus, Mus musculus musculus, and Mus musculus castaneus. Hybrids between mice of the Mus musculus complex are viable and most are also fertile in both sexes. Hybrids of the Mus musculus complex with Mus spretus or Mus spicilegus are viable but male sterile. (Redrawn with permission from Elsevier © 2003, Guénet and Bonhomme 2003.)
Another important observation was that the distribution of high/low SNP rates is unique to a particular pair of strains, when considered on a genome-wide scale. This unexpected heterogeneity found a logical explanation when matching it to historical records about the origin of laboratory-inbred strains and the way these strains were developed during the 20th century. As hypothesized by geneticists, including ourselves in the late 1980s, inbred strains have a polyphyletic origin stemming from three subspecies of the genus Mus: Mus m. domesticus (the occidental wild mice), Mus m. musculus (the oriental wild mice), and Mus m. castaneus (the Asiatic wild mice) (Bonhomme et al. 1987). The genome of a given inbred strain is a unique mosaic, with variable proportions of these three components but with the vast majority of segments deriving from either M. m. domesticus or M. m. musculus (Fig. 3). This is reflected in the genome-wide distribution of SNPs, even if a small proportion of these polymorphisms are of recent origin (mutations). The discovery of these SNPs, which are extremely abundant and very easy to characterize (dense genotyping can be processed automatically), has important implications for the development of several aspects of mouse genetics, including the establishment of pedigrees, the cloning of genes or quantitative trait loci (QTLs), and the comparisons of aligned sequences (Lindblad-Toh et al. 2000; Ideraabdullah et al. 2004).
Where do we go now?
The decision to systematically and comprehensively sequence the mouse genome was certainly a wise decision. There is no doubt that the genome of this important species would have been eventually sequenced one day, but this would probably have been achieved by small “bits,” with many redundancies and, accordingly, at a greater cost for the community. This was also a very democratic decision because laboratories that did not have easy access to sequencing facilities, for whatever reason, can now use this public resource for designing their experiments.
The future of mouse genomics/genetics in the after-sequencing era is relatively clear. The sequence will progressively become complete and precise, and as I have already mentioned, this will aid geneticists who are making interspecific comparisons at high resolution. We will then have another way to understand the forces that shape the genomes. Comparing specific CNSs in a wider panel of species (rat, dog, pig, cattle, Maccaca monkeys, chimpanzee), and matching the results with tissue-specific transcriptomes, will be especially helpful for understanding gene regulation. The design of genetic alterations to be performed in ES cells will also be greatly facilitated by a precise knowledge of the genome sequence. Finally, it seems clear that the genome of several other mouse inbred strains will be sequenced, if not entirely, at least by 0.2- to 1-Mbp nonoverlapping and evenly distributed segments, to enrich the SNPs collection. Comparisons of the SNPs distribution across the genome may be relevant to understanding common polymorphisms, especially those causing strain phenotypic variations and diseases.
Generating many, many new mutations in the mouse genome
Mutations have been collected by mouse fanciers since well before the study of genetics began, and these mutations have been instrumental for the expansion of the discipline. Mouse mutations have also provided, and continue to provide, helpful models to human geneticists. In the post-sequencing era, mutations will still be very useful, contributing efficiently to gene annotation by allowing matching a specific alteration in the DNA sequence to a particular mutant phenotype. Indeed, when a gene becomes nonfunctional after a mutation has occurred, the careful comparison of the mutant phenotype with the normal one, considered together with the molecular defect generated by the mutational event and the spatiotemporal expression of the gene, is an excellent way for assessing the function(s) of the gene in question.
Spontaneous mutations unfortunately have a few major drawbacks. First, they occur at very low frequency (on the average 5 × 10-6 per locus and per gamete) and the number of genotype stored in the repositories of the community, although quite large (around 1200 mutant alleles), was considered by researchers as largely insufficient compared with the expected number of genes in the mouse genome. For example, among the many mutations reported in human that lead to a pathological condition, few have an orthologous counterpart in the mouse and vice versa. This is either because the mutant allele in question never occurred or because it occurred but was not recognized as such and accordingly got lost. To give another example of the shortage of mutant alleles in the mouse, we found that most (over 60%) of the mutations that were discovered spontaneously in our laboratory in the decades 1970 to 2000 turned out to be mutations in a gene that previously had no other alleles rather than new alleles at a locus where mutations already occurred. This clearly indicates that the mouse genome is far from being saturated by mutations, a situation that British colleagues defined as a “phenotype gap.” Another major drawback is that the collection of mutations available corresponds mainly to alleles that are viable ab utero, even if only for a few hours, and have a clear and easily identifiable phenotype. Lethal alleles and those with a weak phenotype or a phenotype with a very late onset (e.g., a hypothetical and eagerly awaited model of Alzheimer's disease) were in general not detected, although they probably represent quite a substantial proportion of all mutations.
To improve the situation, several projects were launched between 1998 and 2005, in different countries, with the aim to generate large amounts of new genetic alterations leading ideally to the production of at least one mutation per gene. Most of these ambitious projects have been extensively described (Durick et al. 1999; Hrabe de Angelis et al. 2000; Nolan et al. 2000, 2002; Nadeau et al. 2001; Hansen et al. 2003; Austin et al. 2004; Auwerx et al. 2004; Clark et al. 2004; Masuya et al. 2004; Schnutgen et al. 2005); we will only briefly outline them here.
Inducing mutations with chemicals
The first initiatives were making use of the powerful chemical mutagen ethyl-nitroso-urea (ENU). ENU induces point mutations (mostly base pair substitutions) and increases the spontaneous mutation rate by a factor of 130 to 160 depending on the experimental conditions (Guénet 2004). Now, at least twelve ENU projects are in progress over the five continents and thousands of mice, offspring of mutagenized progenitors and potentially affected by mutations (dominant or recessive), are carefully analyzed each year for a large number of phenotypic criteria. If we consider the progress reports released by some of the consortia involved in ENU mutagenesis, the strategy appears to be extremely rewarding and many new mutations have been generated this way (Hrabe de Angelis et al. 2000; Nolan et al. 2000; Rathkolb et al. 2000; Clark et al. 2004; Masuya et al. 2004; Hoebe and Beutler 2005; Wilson et al. 2005). These mutations, once identified, are, in general, roughly described phenotypically and then their description is posted on specific websites announcing to the community the availability of new mutant genotypes. Finally, deep-frozen sperm cells are stored to ensure long-term preservation of the genotypes.
The advantages of the strategy making use of ENU are a relatively high efficiency and, mostly, the fact that ENU generates a great variety of mutations (i.e., hypomorphs, hypermorphs, neomorphs, etc.) and not only null alleles. A major drawback of the strategy is that one cannot target the occurrence of mutation hits at a specific site. In fact, mutations occur randomly and, accordingly, characterization of the molecular defect requires the use of the tedious (and expensive) process of positional cloning (forward genetics). Another drawback is that each and every cell of a mouse embryo originating from a mutant gamete (a spermatozoon, in most instances) is affected by the mutation. This may have negative consequences on the survival of the embryo when, for example, the mutant allele impedes normal development for one reason or another. For example, a mutation producing a cleft palate, among other phenotypes, would remain undetected because a baby mouse with such a minor malformation cannot suckle and would die, even if the other tissues of the organism are only weakly affected by the mutation.
The first and most important of these drawbacks has been circumvented by using a clever strategy that consists of the induction of mutations by chemical mutagenesis as usual, and then storing independently DNA samples and spermatozoa from a large number of male G1 offspring of a mutagenized G0 male mouse. In a second step, mutations in a given gene of interest are sought among the collection of G1 DNA samples, using powerful PCR-based high-throughput methods to detect DNA strands mismatches in the targeted region (Coghill et al. 2002; Zan et al. 2003; Quwailid et al. 2004). This region can be a coding sequence or any other sort of sequence of interest, like those coding for RNAs or even the CNSs discussed earlier. INGENIUM, for example, claims that by scoring an archive of some 16,000 G1 male mice (DNAs) they are able to identify over 10 potentially interesting mutations per average-sized gene, and this within only one week. The alleles available in the archive can be evaluated and scored by in silico or in vitro assays for structural or biochemical effects. In a final step, the selected alleles are revitalized from thawed spermatozoa by in vitro fertilization. A resource encompassing over 300,000 unique gene-specific alterations is now available on the C3HeB/FeJ background strain (Augustin et al. 2005). Compared with others, the strategy is fast, dependable, and relatively cheap. In addition, all sorts of mutations are produced, allowing the investigators to choose in an allelic series.
Chemical mutagenesis and radiations have also been used for the massive induction of mutations in vitro in ES cells (Goodwin et al. 2001; Munroe et al. 2004). Here again, the detection of mutant alleles can be performed at the DNA level and the relevant ES cells can then be selected and used for producing heterozygous animals or stored deep frozen for further use. Some of the chemicals and most of the radiations that were used in these experiments produced deletions of various sizes. These deletions, provided they are viable in the heterozygous state, are very precious tools for a detailed analysis of some specific parts of the genome (Rinchik et al. 2002).
Inducing point mutations by genetic engineering in ES cells
As mentioned earlier, chemical mutagenesis is “blind.” This is, at the same time, a blessing and a curse—a blessing because, with this technique of mutagenesis, all sorts of mutant alleles are produced, but a curse because the ambitious aim of producing one chemically induced mutation per gene would certainly be extremely costly if not unattainable. This intrinsic limitation is probably worsened if, as suspected, we consider that chemical mutagens are not equally active on all parts of the genome, leaving some regions less affected than others. Alternatives to chemical mutagenesis have been suggested to complete the “one-mutation-per-gene” challenge. Among these alternatives, the one that consists of making knockouts in vitro, in ES cells, was the first that came to mind.
Engineering knockouts in ES cells is a reverse genetics approach in the sense that one first selects a gene of interest, then makes a specific mutation in it, and finally observes the phenotype of the mutant mouse, if any. To date, it is estimated that knockout alleles potentially exist somewhere throughout the world for only about 10% of mouse genes, but unfortunately many of these knockouts are limited in utility because they have not been phenotyped in standardized ways or are not freely available. However, with the genomic sequence now available, and accordingly the vast majority of genes identified, one could, at least in theory, decide to systematically knock out all the genes in the mouse genome. In addition, considering the wide arsenal of techniques the geneticists now have at their disposition, they could also decide to make all these knockouts conditional and/or tissue specific. To this end, large-scale efforts have recently been launched in Europe (the European Conditional Mouse Mutagenesis Program or EUCOMM project, Auwerx et al. 2004) and in the USA (the knockout mouse project or KOMP project, Austin et al. 2004), and it is likely that other such projects will also be undertaken in other countries with the aim to produce a comprehensive collection of mouse ES cells heterozygous, in some cases homozygous, for a conditional null mutant allele in every gene in the genome. All the mouse strains generated in these projects would then be phenotyped using a battery of sophisticated and standardized tests, including transcriptome-based phenotyping (microarrays), and will finally be made freely available to the community (Schnutgen et al. 2005).
Producing knockout mutations in ES cells with the classical methods is very reliable but rather cumbersome and not easily amenable to high throughput. Gene trapping is an alternative to gene targeting with some interesting advantages. Like homologous recombination, it is performed in ES cells, but, unlike homologous recombination, it is a random approach that produces, with high throughput, a large number of insertional mutations across the mouse genome (Durick et al. 1999; Stanford et al. 2001; Forrai and Robb 2005). Gene trapping disrupts genes by inserting an engineered DNA element (a promoterless reporter/selectable cassette flanked by an upstream 3′ splice acceptor and a downstream adenylation sequence). When the DNA vector inserts in an intron of an endogenous gene and the gene is expressed, a fusion mRNA is transcribed to produce a nonfunctional version of the cellular protein fused to the reporter/selectable marker. The gene trap strategy has then three advantages over other techniques: (1) it inactivates the genes randomly; (2) it reports the expression of the trapped gene when the latter is expressed; and (3) it provides a DNA tag for PCR identification of the disrupted gene. This approach has been used by a few laboratories, members of the former Gene Trap Consortium, to generate a public resource of roughly 36,000 characterized ES cells, harboring null mutations in approximately one third of the genes (10,000) (Hansen et al. 2003; Stryke et al. 2003; Skarnes et al. 2004). The resources generated by this consortium, although very abundant and very useful, suffer from a drawback that I have already mentioned: They do not produce conditional alleles and, accordingly, the analysis of mutants generated from these resources is limited to the earliest developmental functions of the trapped gene. The aim of the EUCOMM consortium is to generate, in the five years to come, a library of up to 20,000 different engineered ES cells strains, with roughly 12,000 of these cells being randomly gene trapped (Adams et al. 2004; Austin et al. 2004; Schnutgen et al. 2005). This initiative is extremely interesting because, unlike the former program and thanks to a clever design of the trapping vector, the new mutations will be conditional. The mutations in the new library will enable analysis of gene function(s) in a temporally and spatially restricted manner and will be either in the germ line or in somatic cells/tissues, depending on the deletion system used. In addition to the massive random production of inactivated genes by gene trapping, some other genes (8000), selected a priori for their relevance to human pathology and that may have been missed by the gene trapping project, will be inactivated by gene targeting using a new kind of insertional targeting vectors with high targeting efficiency (Adams et al. 2004). Here again, the mutant mice generated will be thoroughly phenotyped, archived, and made available to the public. Using the two strategies is interesting because they are really complementary. Gene trapping is fast and very efficient but, as mentioned above, those genes that are not expressed in vitro, in ES cells, would not be “trappable,” but targeting could inactivate all these genes.
Based on what we described in the previous lines, the collection of mouse mutations may turn plethoric in the forthcoming years. If we add the number of chemically induced mutations, in vivo or in vitro, to those resulting from the complementary efforts of the “gene trappers,” we can seriously expect a vast majority of the mouse genes to have at least one mutant allele (in most instances a knockout) in the next five years. Mouse geneticists may then feel like they are in Wonderland!
Just like for the sequencing of the genome, the decisions to provide funding for the projects aimed at systematic mutagenesis of the mouse genome were also wise decisions. These projects will undoubtedly contribute to a greater efficiency in modern genome research and, in the end, they will also save a lot of public funding that could be put on other important themes. Many laboratories with very little or no experience in ES cells technology will certainly appreciate being able to order a conditional knockout allele of their favorite gene.
Unfortunately Wonderland does not exist, and many genes will escape chemical mutagenesis or gene trapping. The efficiency of chemical mutagenesis depends, in part, on the size of the gene and, for this reason, the very small-sized genes will be difficult to hit. In the same way, those genes with only one exon, which are so far mostly unknown, won't be “trappable.” Mutant alleles with a cellular dominant lethal effect will be extremely difficult, not to say impossible, to analyze. Another important point is that, while only one null allele can be induced per gene, an infinite number of alleles resulting from missense mutations are possible. Replacing any base pair with one of the other three may, in some cases, lead to amino acid substitution with totally unpredictable consequences in the protein. We found, for example, that the mouse mutation pmn, which is a Trp524Gly substitution in the sequence of the chaperone molecule TBCE (Tubulin folding co-factor E), resulted in a severe neurological syndrome with relatively late onset, whereas a 2-bp deletion, which probably inactivates the orthologous human gene, leads to a totally different pathology (Kenny Caffey syndrome OMIM 244460 or Sanjad-Sakati syndrome; OMIM 241410) (Martin et al. 2002). It is presumably because the mutant protein is unstable in the mouse and absent in humans that the phenotype is so different. In this case at least, analysis of a null allele of the Tbce gene would have been insufficient for the complete annotation of the functions of this gene. This clearly indicates that spontaneous mutations with interesting and unique phenotypes will always be interesting to consider for positional cloning. Finally, the mutations that can be induced in the CNSs are innumerable, and so far, we have no way of predicting their effects and very few strategies for generating them with high throughput.
Engineering chromosomal rearrangements in mice
As discussed above, the modern techniques of ES cell technology have revolutionized our ability to produce mouse genomic alterations and point mutations in particular. They also have permitted us to produce a virtually unlimited number of chromosomal rearrangements that will be very useful either as tools for mouse geneticists or, maybe more importantly, for modeling human diseases. Using the Cre/loxP site-specific recombination system and the knowledge we now have of the mouse genome sequence, one can very precisely delete, duplicate, or invert chromosomal segments of various size, almost at will, just by inserting the appropriate number of loxP sequences at specific sites, in cis or in trans, and in the appropriate orientation (Yu and Bradley 2001). For the production of deletions there is a limit (in particular in size), which is related to the impossibility of putting some chromosomal segments in the haploid state. Deletions and nested deletions can be produced that are useful for generating resources for regional recessive genetic screens and for facilitating the functional analysis of the mouse genome (Nobrega et al. 2004). Deletions and duplications are useful for studying the effect of gene dosage on cell physiology because any gene in the mouse genome can be put in different copy numbers (Yan et al. 2004). Chromosomal engineering can also be used to generate duplications in mouse chromosomal regions conserved with the trisomic human chromosome 21 regions to identify the genetic domain(s) and causative gene(s) that are responsible for the clinical characteristics of the Down syndrome (Reeves et al. 2001). This strategy is an alternative to the one of targeted meiotic recombination (TAMERE) but seems to be more efficient (Herault et al. 1998; Olson et al. 2005). Inversions are useful because they are cross-over suppressors and allow the creation of balancer chromosomes (Kile et al. 2003).
Reciprocal translocations can also be engineered in ES cells and can be produced to develop mouse models for certain forms of human cancer resulting from the creation of novel fusion genes with cellular oncogenes.
The phenotyping programs and the genetic analysis of complex traits: A challenge for the future
The sequencing of the mouse genome and the massive production of new mutations have been, undoubtedly, the two most important projects of the last decade in mouse genetics, and I already have emphasized that these two projects were indeed complementary in the sense that, while sequencing identifies the genes through their DNA structure, mutations identify the genes through their function and contribute to gene annotation. The development of these two projects has triggered other initiatives and I would like to mention two other projects, which are also complementary and will probably mark an important change in the analysis of gene functions. The aim of the first project is to perform accurate and comprehensive phenotypings of the different inbred and mutant strains. The aim of the second is to develop new tools and strategies for the genetic analysis of heritable traits with a complex or multigenic determinism.
The phenotyping programs
The Mouse Phenome Project was created in May 1999, with the aim to establish a collection of baseline phenotypic data of the mouse inbred strains (Paigen and Eppig 2000). In a first step, the most commonly used inbred strains have been classified in four groups by a panel of specialists, and for each group, reliable phenotypic data are now being progressively collected and stored in a central, web-accessible database (the Mouse Phenome Database or MPD), housed at The Jackson Laboratory and cross-linked to the Mouse Genome Database. This large-scale collaborative project, mainly based on the free, willing participation of expert scientists in diverse fields of biomedical science, was another wise decision because any laboratory embarking on a new research project can now select, in only a couple of “clicks,” the best strains to work with based on the existing collection of phenotyping data. As I will discuss further, the creation of this database will progressively become an essential resource for realizing the full utility of information that emerged from the sequencing of the mouse genome.
Since the foundation of the Phenome database, other initiatives with the same aim of accurate phenotyping have been or will be developed in several countries, in particular, in Europe and North America. EUMORPHIA, for example, is a consortium of 18 research institutes from across Europe that is developing a comprehensive, robust, and validated phenotyping platform in which mutant strains will be thoroughly scrutinized, using a broad range of state-of-the-art technologies for detecting even the most subtle phenotypic changes. The new screen, known as EMPReSS (European Mouse Phenotyping Resource for Standardized Screens), will incorporate more than 150 standard operating procedures, covering all of the main body systems (Auwerx et al. 2004; Brown et al. 2005). These mouse “phenotyping workshops” are, of course, logical corollaries of the projects aimed at the induction of new mutations and represent enormous progress for a better characterization of gene functions. After accurate and comprehensive phenotyping, it is likely that many knockouts that were generated in the last twenty years and so far have been classified as “normal” (i.e., with no detectable phenotype and accordingly useless for genome annotation) will be found to be “abnormal” on a second examination with more refined protocols. It would be even more interesting if, in the future, all these workshops or “clinics” could develop a network of exceptional expertise and work on an integrated basis to resolve the difficult cases. This would save a lot of money but would require that interesting genotypes be freely and easily exchanged worldwide, with these exchanges not being impeded by sanitary or legal issues. This point should be seriously considered by the community.
The genetic analysis of complex traits
Until now, and in the vast majority of cases, correlations between a particular phenotype (generally one with deleterious aspects) and its determinism at the genome level have been established only for monogenic traits. Positional cloning of a mouse mutation is a typical example in which a single Mendelian character determines a pathology and a causal relationship can be established by finding a correlated alteration in the sequence. Many such genes have been cloned and, presumably, many more will be cloned in the years to come, given that, after the sequencing of the genome, the strategy is greatly facilitated and, after the massive production of new alleles, many interesting phenotypes that were missing will be available. The problem however is that most of the pathologies are not “monogenic” but, on the contrary, are influenced by multiple genes with additive or synergistic effects. In the same way, most alterations that have been found to account for a deleterious phenotype in the mouse have been found to affect the coding regions of the mutated gene (base pair substitutions, deletions, insertions, splicing abnormalities, etc.), but mutations with an effect on the quantitative or spatiotemporal expression of a gene are not well known although they are probably quite common. Finally, many genes with a modifier effect, increasing, for example, the severity of a phenotype or making a certain inbred strain of mouse more or less susceptible to an infectious disease or a certain type of cancer, have been identified only exceptionally. In fact, in our analysis of the genotype/phenotype relationships so far, we have probably considered only the “tip of the iceberg,” because we have had no tools suitable for assessing the genetic analysis of complex traits. As a consequence of the mouse genome sequencing effort, and in particular after the discovery of so many SNPs in the various inbred strains, and as a direct consequence of the development of better phenotyping strategies, one can expect the situation to change dramatically in the forthcoming years. An extremely dynamic consortium dealing with all these problems, the Complex Trait Consortium or CTC, was created in the spring of 2002, by a group of expert scientists who decided to identify research priorities and tools to tackle the problems related to quantitative inheritance in the mouse model. The goals of the consortium have been published in several scientific journals (Glazier et al. 2002; Abiola et al. 2003; Nishimura et al. 2003; Churchill et al. 2004; Pletcher and Wiltshire 2004; Singer et al. 2004). In short, they are preparing and collecting the tools that would allow relating specific haplotypes (or segments) of the mouse genome, identified by a particular set of SNPs, with a particular phenotype identified by a QTL (Petkov et al. 2004; Pletcher et al. 2004). Among the strategies suggested by the CTC, the most impressive and certainly the most innovative is the implementation of a resource known as Collaborative Cross (Churchill et al. 2004). The Collaborative Cross will consist of a total of 1000 recombinant inbred strains (RIS), each derived from an initial eight-way cross involving very different and unrelated inbred strains (Fig. 4). Theoretical computations indicate that the genome of each RIS in such a cross will capture ∼135 unique recombination events (135,000 for the whole set of RIS) and each of these RIS will then have a unique genomic constitution representing a patchwork of 135 elements with, roughly, an equal proportion of the eight founder genotypes. Each strain of the Collaborative Cross will capture an abundance of polymorphisms every 100-200 bp that will be sufficient to drive phenotypic diversity in almost any trait of interest, provided it segregates among the eight parental strains. Finally, the very large number of RIS will guarantee high mapping resolution (achieved by SNPping) of any QTL segregating in at least two of the eight parental strains.
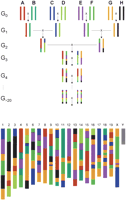
The collaborative cross stems from an 8-way interstrain cross. Eight carefully selected inbred strains are crossed to produce four F1 hybrids. These hybrids are then crossed together; one thousand independent inbreds are derived after 20 additional generations of brother × sister mating. In the end, this cross will be materialized by a resource of about one thousand different inbred strains (actually recombinant inbred strains, RIS) whose genome will be a “patchwork” with a roughly equivalent contribution of the original inbred strains. For each strain the “patchwork” will be unique. Altogether, the resource of 1000 strains will represent 135,000 recombination events in the mouse genome and will segregate for a large quantity of polymorphisms. (Redrawn with permission from Nature © 2004, The Complex Trait Consortium, 2004.
The two projects discussed above—the phenotyping programs and the analysis of complex traits by the partners of the CTC—are really innovative. Unlike the projects aimed at sequencing the mouse genome and those aimed at the production of new mutations, they have not yet reached their “full speed,” since the essential tools are not yet available, but there is no doubt that with these research programs we will certainly have a better idea of quantitative inheritance. Even if the eight strains that have been selected as founder strains of the eight-way cross represent only a sample of the polymorphisms that may segregate in the mouse species, this will probably be more than enough to allow unraveling of at least some elementary mechanisms of quantitative inheritance in mammals. Another conclusion we must draw from these last two initiatives is that they both were the consequence of a very high level of interactive and constructive consultation inside the community. This is a big change in the mentalities to the benefit of the progress in mouse genomics.
Conclusions
The aim of this review was to summarize the most important advances in mouse genetics that occurred over the last fifteen years. Among these advances, the release on public databases, in 2002, of a first draft of the mouse genome sequence is certainly the one that had the greatest impact on the community. The other projects described that are aimed at understanding the function(s) of genes or the complex nature of quantitative inheritance will certainly also influence dramatically the future of mouse genetics and change the way we have been approaching human health and disease.
This review, however, is not comprehensive, and many other subjects would also have deserved to be discussed. For example, the extraordinary progress made by molecular geneticists for engineering alterations in ES cells should be mentioned. While homologous recombination was only in its infancy fifteen years ago, it has now reached a very high level of sophistication with the possibility of making, almost at will, a conditional or inducible and tissue specific mutation in each and every gene (Kos 2004). The advent of siRNA-directed technology for gene silencing, which is only in an early step of development at the moment but will certainly play an important role in the arsenal of mouse geneticists in the future, is also worthy of mention (Hasuwa et al. 2002). Finally, I should mention the development of new inbred strains of mice, derived from recently trapped wild specimens of the same genus Mus but from different species or subspecies. These strains offer geneticists a virtually unlimited amount of polymorphisms of all kinds and an endless variety of new alleles that have been selected by chance (or by necessity?) under the only pressure of natural selection (Guénet and Bonhomme 2003; Ideraabdullah et al. 2004). It would be interesting, for example, to set interspecific crosses and study the consequences of bringing together, in the same cell, the products of genes separated by divergent evolution. This could help to identify genetic functions that are subject to rapid divergence and may help to pinpoint the functions that eventually promote speciation. Questions concerning epistatic interactions may also find an answer by analyzing the phenotype of offspring of interspecific crosses at the genomic level and assessing the consequences of “packing” into the same genome alleles stemming from distantly related species. So far, we have no clear answers to these questions, but data exist indicating that some combinations of alleles are strongly counterselected in the offspring of some interspecific crosses (Montagutelli et al. 1996) and deleterious phenotypes, such as diabetes, autoimmune diseases, or male sterility, are common. Our attempts to develop interspecific consomic (or chromosome substitution) strains with complete chromosomes of Mus spretus introgressed into a C57BL/6 background substituting for the original chromosome have failed in most instances, while consomic strains have been easily created between strains of the same species (Singer et al. 2004). Again, we have no explanation for this, but deleterious epistatic interactions due to genetic divergence from the ancestral alleles are highly suspected.
As discussed, the last fifteen years have obviously been extremely important, providing the community with an enormous amount of new information, but what is also remarkable is that all of this information and these tools, thanks to the Internet, have been made available to the public very rapidly and at no cost. Then, if the microcosm of mouse geneticists has changed so dramatically (and for the better!) over the last fifteen years, maybe it is time to thank the legion of anonymous curators taking care of all these databases. They have made the world a bit better.
Acknowledgments
I thank Professor Steve D.M. Brown, for reading this manuscript and making interesting suggestions. This review is dedicated to Professor François Jacob who, 35 years ago, convinced me as a young vet that the mouse was really an interesting animal model for geneticists. It is also dedicated to the scientists at Harwell and Bar Harbor who gave generously of their time to teach me the basics of Mouse Genetics.
Footnotes
-
E-mail jlguenet{at}wanadoo.fr; fax 33 1 45 68 86 34.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3728305.
- Cold Spring Harbor Laboratory Press








