
Drosophila melanogaster: A case study of a model genomic sequence and its consequences
Abstract
The sequencing and annotation of the Drosophila melanogaster genome, first published in 2000 through collaboration between Celera Genomics and the Drosophila Genome Projects, has provided a number of important contributions to genome research. By demonstrating the utility of methods such as whole-genome shotgun sequencing and genome annotation by a community “jamboree,” the Drosophila genome established the precedents for the current paradigm used by most genome projects. Subsequent releases of the initial genome sequence have been improved by the Berkeley Drosophila Genome Project and annotated by FlyBase, the Drosophila community database, providing one of the highest-quality genome sequences and annotations for any organism. We discuss the impact of the growing number of genome sequences now available in the genus on current Drosophila research, and some of the biological questions that these resources will enable to be solved in the future.
It is almost 100 years since William Castle introduced Drosophila melanogaster to the pleasures and rigors of biological research (Castle 1906). Four major phases of Drosophila research can, perhaps, be distinguished. The period ∼1910-1940, of classical genetic analysis was a period of rapid development in which most of the major principles of classical genetics were established: the chromosome theory of heredity, the nature of genetic linkage and genetic maps, the genetic behavior of chromosome aberrations, the induction of gene and chromosome mutations by radiation, the discovery of mitotic recombination, and so on. This was followed by a long period, ∼1940-1968, of growth but relative sterility, a period in which many of the best minds in genetics turned their attention to microbes and phage. The period from, roughly, ∼1968-2000 was a renaissance, witnessed by many molecular biologists moving into the field, creating an analytical, rather than descriptive, study of development and behavior. This metamorphosis was fueled by many major technical advances within the field, for example, the invention of in situ hybridization, of the P-element-based transformation technology, of powerful methods for clonal analysis, the discovery of potent chemical mutagens, and by the extraordinary external advances in molecular biology. New generations of researchers selected Drosophila as a model organism for the study of fundamental problems in biology. From 2000, fly research has matured into its fourth period: the genome era, for, on March 24, 2000 the first release of the “complete” genomic sequence of Drosophila melanogaster was published, timed to coincide with that year's annual fly meeting in Pittsburgh. Five years into the post-genomic era we can begin to ask: What have we learned and what may lie ahead?
The genome
Prior to 1998, two groups, the Berkley Drosophila Genome Project and the European Drosophila Genome Project, were beginning to sequence the genome of D. melanogaster by the tried and tested way of sequencing a minimal tiling path of clones (cosmids, P1 clones, and BACs) chosen from physical maps of the genome (Hartl et al. 1992; Madueno et al. 1995; Kimmerly et al. 1996; Hoskins et al. 2000). That changed on May 12, 1998, when Craig Venter invited Gerry Rubin to participate in an attempt to sequence the genome of this fly by whole-genome shotgun sequencing (WGS), a method untried and untested for anything larger than a bacterial genome of one or a few megabases in size. There was considerable skepticism in the community that WGS would succeed for a large and complex genome with much repetitive DNA (see Green's 1997 riposte to Weber and Myers' 1997 paper in this journal). It was a leap of faith that the combination of the new capillary sequencing machines, of very careful construction of clone libraries, and of software (then not yet written) would allow the 120-Mb euchromatic genome of D. melanogaster to be assembled. By September 1999 this faith had been justified: A WGS assembly of the euchromatic portion of the fly genome had been achieved. This proof-of-principle for a metazoan WGS was the first landmark contribution of the fly genome project.
At that time, only one metazoan genome, that of Caenorhabditis elegans, had been sequenced and annotated (The C. elegans Sequencing Consortium 1998). Experience in genome annotation, both as a technical problem and as a community problem, was sparse. The “annotation jamboree,” hosted by Celera in November 1999, was important, not only for what it did, but for how it did it—an intimate and intensive collaboration of software engineers and of biologists drawn from the community working together both to build gene models and to annotate these with functional information using the then fledgling Gene Ontology (http://www.geneontology.org/) (Ashburner et al. 2000; Lewis 2005). This is a model that has been followed by many other communities. Also unusual (although not unique, vide C. elegans) to this day was the very close association between the sequencing and annotation groups and FlyBase (http://www.flybase.org/), the community database for Drosophilists. The act of analyzing the fly genome sequence, therefore, was novel in a second respect: It introduced new community methods for genome annotation and curation.
The “complete” sequence of the genome of D. melanogaster we have today is not that released in March 2000 (Adams et al. 2000; Myers et al. 2000; Rubin et al. 2000). Since Release 1, there have been three subsequent genome releases (Celniker et al. 2002) (Release 4 was in April 2004; Release 5 is planned to be the final release of the genome sequence); each release is improved in quality, with the correction of errors, both of sequence and assembly, the closure of physical and sequence gaps (only 23 now remain in the Release 4 euchromatin), and the correct assembly of repetitive sequences (this has been the responsibility of the Berkeley Drosophila Genome Project; http://www.fruitfly.org/). In addition, a separate project, the Drosophila Heterochromatin Genome Project, has been funded to sequence the complex heterochromatic sequences of the telomeres and pericentromeric chromosome regions (http://www.dhgp.org/). FlyBase has been responsible for keeping the annotation of the genome up to date. A major effort by about 10 FlyBase annotators resulted in a complete revision of all gene models and other genome features, based on the first “finished” (Release 3) sequence; this was published in a series of papers in a special issue of Genome Biology in December 2002 (http://www.genomebiology.com/Drosophila). Revision of gene models and other features subsequently is an ongoing, reiterative, task being done by FlyBase (Drysdale et al. 2005).
Immediate lessons
Before November 1999 there had been decades of debate as to the number of protein-coding genes in D. melanogaster. That debate then stopped: it is ∼14,000. Some, for example, Hild et al. (2003), have argued that the number of protein-coding genes had been seriously underestimated (perhaps by as many as 2000 protein-coding genes) by the original annotation. A careful experimental evaluation of these “missed” gene models shows few of them to be real; many are simply new exons of genes already known or predicted (see Yandell et al. 2005). Before December 2002, the abundance and diversity of the transposable elements in the genome of D. melanogaster was unknown: The first attempt at their annotation (Kaminker et al. 2002) gave numbers of 1572 elements in 93 families; a more recent analysis using improved methods and including additional families (such as the enigmatic INE-1 element) (Locke et al. 1999), indicates that the Release 4 “euchromatin” (an operational definition for the assembled chromosome arms including the first few megabases of the pericentromeric heterochromatin) has 6013 elements in 127 families (Quesneville et al. 2005; http://dynagen.ijm.jussieu.fr/repet/dmel4/index.html).
Added benefits
In addition to revealing the parts list of the Drosophila genome, the completed sequence of D. melanogaster has changed the practice of Drosophila genetics and led to many unexpected discoveries. Having the genome has enormously accelerated—by a factor of at least 10—the time required to clone a particular gene of interest; this tedious task is no longer rate limiting or essential for biological discovery. The large, and growing, collection of inserted transposons used for gene disruption (mostly P-elements, but also hobo, Minos, and piggyBac) can now be mapped precisely to the genome sequence, rather than to a 50-100-kb interval by in situ hybridization to polytene chromosomes. About 65% of the genes of D. melanogaster have been disrupted by at least one transposon insertion (Bellen et al. 2004; Thibault et al. 2004; Venken and Bellen 2005). With advances in P-element technology, this has led to methods for the construction of deletions whose limits are known with base-pair accuracy, and to attempts to cover the entire genome with a minimal tiling path of deletions (Parks et al. 2004; Ryder et al. 2004). Single nucleotide polymorphisms between the sequenced strain and others have led to the construction of several SNP maps, which enormously help the mapping of, for example, EMS-induced point mutations (Berger et al. 2001; Hoskins et al. 2001; Martin et al. 2001). The genome sequence has also greatly facilitated the recovery of EMS-induced mutations in selected gene regions using the method of tilling (Winkler et al. 2005).
The completion of the fly genome in 2000 coincided with great advances in genomic technology that have revolutionized our abilities to study transcription, protein binding to specific DNA sequences, and genetic variation at the molecular level. We can now make microarrays for expression profiling, either targeted to all known or predicted coding regions or against wholegenome tiling paths of high resolution (e.g., the INDAC resource; see http://www.indac.net/); we can now map the binding sites of chromatin-associated proteins to the genome at high resolution, using either DamID (Orian et al. 2003; Sun et al. 2003; Bianchi-Frias et al. 2004) or chromatin immunoprecipitation (chIP) (MacAlpine et al. 2004; Birch-Machin et al. 2005); we can now conduct genome-scale surveys for polymorphisms using high-throughput PCR strategies (Glinka et al. 2003; Orengo and Aguade 2004), and effectively re-sequence other genomes of the same species, using tiling paths of oligonucleotides (http://www.dpgp.org/). Genome resources have also revolutionized the genetic studies of complex traits in Drosophila (Pletcher et al. 2002; Harbison et al. 2005).
The task of obtaining one full-length cDNA from each fly gene is not only facilitated by the genomic sequence (Stapleton et al. 2002a,b), it helps enormously in refining gene models (Misra et al. 2002). We can now look forward to the day when each gene is represented by one full-length cDNA (in a versatile vector that will allow it to be shuttled to a variety of useful constructs) (http://www.fruitfly.org/EST/; S. Celniker, pers. comm.), and perhaps even to the availability of full-length cDNAs from every alternatively spliced transcript. Likewise, the genomic sequence has enabled the design of antisense RNA reagents that are now allowing large-scale, systematic RNAi screens for gene function in tissue culture cells (Bettencourt-Dias et al. 2004; Boutros et al. 2004).
The proper study of the genome is the genome itself. Quite unexpected properties of genomes have come from following this edict. Many individual examples of tandemly repeated genes had been known from work prior to the genome. But it was only the analysis of a 2.9-Mb trial sequence (Ashburner et al. 1999) and of the genome itself in 2000 that showed just how common this is, and the extent to which some protein families (e.g., of serine proteases) had expanded by duplication. Similarly, nested genes were first discovered in flies (Henikoff et al. 1986), but these were thought to be rare exceptions: They are not. More than 7% of the genes in the D. melanogaster genome are nested (Ashburner et al. 1999; Misra et. al 2002), and flies have at least a dozen examples of nests within nests. mRNAs that do not encode proteins also appear to be more common than previously thought (Tupy et al. 2005), an observation that may help to explain the phenomenon of “intergenic” transcription (Hild et al. 2003; Stolc et al. 2004).
The analysis of the genome of D. melanogaster has led to the insight that this genome is far more complex than we had imagined. In flies, as in other species (Cohen et al. 2000; Caron et al. 2001; Roy et al. 2002), the sequence has allowed us to observe that the genome is organized into large gene-expression neighborhoods, within which even unrelated genes tend to be coexpressed (Boutanaev et al. 2002; Spellman and Rubin 2002; Parisi et al. 2004; Stolc et al. 2004; Belyakin et al. 2005; Thygesen and Zwinderman 2005). The existence of gene-expression neighborhoods suggests coadapted genomic regions that may be related to chromatin domains that may be preserved as syntenic regions during evolution (see below). The strongest evidence for gene-expression neighborhoods appears to come from genes expressed in the male testis (Boutanaev et al. 2002; Parisi et al. 2004), a genomic organization that may be necessary to facilitate proper gene expression during the final stages of sperm development in a highly condensed chromatin environment. The distribution of intergenic lengths in the compact D. melanogaster genome has also been shown to be nonrandom: Genes with complex regulation have long intergenic regions (Nelson et al. 2004). These observations suggest that relationships between genome structure and gene regulation are encoded in the fly genome sequence.
It is no coincidence that perhaps the greatest recent breakthrough in our understanding of gene regulation has come after the completion of genomic sequences of key eukaryotes like Drosophila: the discovery of the vast array of microRNAs (miRNAs) and their functions. In fact, the genome sequence of D. melanogaster helped reveal the fundamental hairpin structure of premiRNAs from mature miRNA expressed sequences (Lagos-Quintana et al. 2001). In turn, this detailed understanding of miRNA structure has allowed their genome-wide prediction in Drosophila (Lai et al. 2003). The genome sequence has also been critical for the prediction of miRNA targets (Enright et al. 2003; Stark et al. 2003; Rajewsky and Socci 2004; Brennecke et al. 2005; Burgler and Macdonald 2005).
There is still much to do
We hope not too many scientists will think that all the fun is over with Drosophila, and turn to the study of the Trichoplax or Loxodonta genomes. There remains much to discover, and many resources are now available to catalyze discovery by individual research groups (Matthews et al. 2005), who will remain the bedrock of the Drosophila community in the post-genomic era (Gilbert 1991). Large-scale projects to catalog functional elements in the genome sequence could be integrated and distributed through a Drosophila ENCODE project (http://rana.lbl.gov/drosophila/dencode.html), which would capitalize on the tradition of resource-sharing among Drosophilists, and serve as a model for community-driven, comprehensive genome annotation in higher eukaryotes.
The genomic sequences of a further 11 species of Drosophila (http://species.flybase.net/) will provide a rich source of data for expanding on lessons learned from the D. melanogaster genome. Drosophila genome sequences may, in fact, continue to push advances in WGS and comparative assembly techniques by providing in D. melanogaster a “finished” reference genome. The impact of finishing on genome assembly, annotation, and biological inference can now be evaluated to direct future strategies for genome sequencing projects (Myers et al. 2000; Benos et al. 2001; Celniker et al. 2002).
Heterochromatin has long been recognized as a major, yet mysterious, component of most metazoan genomes. We have already learned much about its molecular nature from studies with Drosophila (Dimitri et al. 2005). We know, for example, that much of the complex heterochromatin of D. melanogaster is composed of a graveyard of decaying, often nested, transposable elements with a sprinkling of protein-coding genes (Hoskins et al. 2002; Dimitri et al. 2003). We know that its chromatin differs in the spectrum of its proteins (Elgin and Grewal 2003). But, it would be an exaggeration for even the most zealous “heterochromatist” to claim that we have anything approaching a full understanding of either the structure or function of this important genome component. The completion of the sequence of D. melanogaster now requires the sequence of the complex heterochromatin (we except the 36 Mb of simple sequence satellite sequences) and new methods for its analysis.
Straightforward in principle, but demanding in practice, is the challenge to discover “functions” for all of the genes. The Gene Ontology has provided not only a structured language to describe gene “function,” but also tools for the prediction of gene function. Yet no scientist should be satisfied for long with only predicted function. Of the 14,461 predicted protein-coding genes of D. melanogaster, only 5402 have known mutant alleles; on the other hand, there are 9875 genes in D. melanogaster whose existence is reasonably well attested by classical methods but that have yet to be identified on the sequence (data computed from FlyBase) (A. de Grey, pers. comm.). Linking the wealth of results published in the literature to the genome is absolutely necessary if we are to leverage the depth of our understanding of development, behavior, and evolution in Drosophila using the genome sequences. Continued progress toward completion of the gene disruption projects and expression profiling (see above) will prove essential for finding functions for the remaining as-yet-uncharacterized genes.
Progress, both experimental and computational, in the understanding of regulatory networks in Drosophila is dramatic: Indeed, it can be argued that the regulation of A-P and D-V axes formation in early fly development is one of the best (if not the best) understood complex biological system (http://bdtnp.lbl.gov/). The syncytial embryonic environment is also optimal for the decoding of networks based on transcriptional control. Indeed, these networks can even be emulated in vitro (Isalan et al. 2005). Yet, from experimental analyses, we understand in any detail the structure of the regulatory regions of relatively few genes, and the annotation of even this limited set is regrettably incomplete (http://www.flybase.org/annot/dmel_release4.1.txt). Nevertheless, the ability to reconstruct core features of the Drosophila segmentation network automatically in silico from annotated regulatory sequences (Fig. 1) suggests that a complete genomic inventory of regulatory elements will have direct impact on gene regulatory network analysis in flies. The genomic sequence will enable great advances here, as computational methods for the prediction of regulatory regions (Berman et al. 2002; Ohler et al. 2002; Rajewsky et al. 2002; Ochoa-Espinosa et al. 2005), particularly using comparative data (Bergman et al. 2002; Berman et al. 2004; Grad et al. 2004; Sinha et al. 2004), improve and as the size of the database of functionally characterized cis-regulatory sequences mapped to the genome increases (Lifanov et al. 2003; Bergman et al. 2005). The development of a genome tile microarray in Drosophila will also be essential for experimentally enumerating the targets and binding specificities of the vast majority of the >700 predicted transcription factors in the fly genome, for which no regulatory information is currently available. Integrating results of gene networks inferred from multiple genomic and proteomic (e.g., two-hybrid screens) (Giot et al. 2003; Stanyon et al. 2004; Formstecher et al. 2005) data sources will hopefully expand and link together functional regulatory modules into coherent systems that specify fly biology.
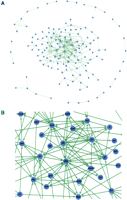
Partial gene regulatory network (GRN) for Drosophila melanogaster automatically generated from genome annotations. (A) GRN determined from entire set of protein-DNA interactions in the Drosophila DNase I footprint database (http://www.flyreg.org/) using Cytoscape (Shannon et al. 2003). Note that the majority of currently annotated protein-DNA interactions coalesce into a single interconnected network. (B) Sector of GRN shown in panel A demonstrating that known transcriptional regulatory interactions can be computed directly from text-based annotation of genomic DNA.
We doubt that the discovery of gene-expression neighborhoods is the last surprise for our understanding of genome structure at a large scale. Here, we believe that comparative data will have much to say. It was about 70 years ago that Sturtevant and Dobzhansky discovered that overlapping inversions can be used for phylogenetic reconstruction (Sturtevant and Dobzhansky 1936), a fact most remarkably used to study the relationships for the >120 species of endemic Hawaiian species (Carson et al. 1992). Now, the high rate of genome restructuring by inversions in Drosophila (Ranz et al. 2001) can be used to reveal groups of genes that maintain positional order, perhaps for functional reasons such as coexpression. The relationship between syntenic regions and gene expression neighborhoods is only beginning to emerge (Stolc et al. 2004); however, the genomic material to reveal the scope of functional constraints on chromosome restructuring is now available. The rigorous definition of syntenic regions and the development of software to achieve this end are an important proximate goal, but the vagaries of incomplete genome sequences, overlapping inversions, and the possibility of breakpoint re-use will present challenges to reconstructing the complete history of inversions in Drosophila.
One of the great lessons of the post-genomic era is the added value of comparative sequence data for the functional annotation of model systems such as Drosophila. The second genome in the genus, that of Drosophila pseudoobscura, was published in January 2005 (Richards et al. 2005) and represents the first stage in the explosion of comparative genomics data being generated currently in flies (http://rana.lbl.gov/drosophila/multipleflies.html), an inevitability heralded by the D. melanogaster WGS. As with interpreting all biological systems, the D. melanogaster genome must be viewed as the product of evolution; thus, the decoding of functional information in this model genome will be intimately intertwined with knowing the evolutionary history and forces that have produced it. Much, however, remains to be learned about the mechanisms of genome evolution in Drosophila: the relative contributions of gene transposition and inversions to gene movement (Ranz et al. 2003b); the gain of lineage-specific genes by retrotransposition and tandem duplication (Betran et al. 2002; Wang et al. 2004); the divergence and proliferation of transposable elements (Kaminker et al. 2002; Lerat et al. 2003; Sanchez-Gracia et al. 2005); the evolution of cis-regulatory sequences (Emberly et al. 2003; Phinchongsakuldit et al. 2004; Ludwig et al. 2005; Negre et al. 2005; Sinha and Siggia 2005); the relationship between cis-regulatory and transcriptome evolution (Ranz et al. 2003a; Rifkin et al. 2003); and the resolution of the roles that mutation, recombination, genetic drift, population history, and natural selection have jointly played in shaping the genomic landscape we observe today (Andolfatto 2001; Aquadro et al. 2001; Glinka et al. 2003; Orengo and Aguade 2004; Haddrill et al. 2005). By identifying both highly conserved and positively selected sequences to predict and inform function, the integration of evolutionary genomics with classical and forward genetics will continue to propel biological discovery long into the second century of fly research.
Acknowledgments
We thank Brian Oliver, Nipam Patel, Gerry Rubin, and two anonymous reviewers for comments on the manuscript of this review. We thank Hamid Bolouri for showing us the potential of Cytoscape. We apologize for any oversight in attribution resulting from space limitations. C.M.B. is supported by a USA Research Fellowship from the Royal Society. Work in M.A.'s laboratory is supported by an MRC Programme Grant to M.A. and Steve Russell.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3726705.
-
↵2 Corresponding author. E-mail ma11{at}gen.cam.ac.uk; fax 44-1223-333992.
-
↵1 Present address: Faculty of Life Sciences, University of Manchester, Manchester, M13 9PT, United Kingdom.
- Cold Spring Harbor Laboratory Press








