
Grains of knowledge: Genomics of model cereals
Abstract
The economic and scientific importance of the cereals has motivated a rich history of research into their genetics, development, and evolution. The nearly completed sequence of the rice genome is emblematic of a transition to high-throughput genomics and computational biology that has also pervaded study of many other cereals. The relatively close (ca. <50 million years old) relationships among morphologically diverse cereals native to environments that sample much of global geographic diversity make the cereals particularly attractive for comparative studies of plant genome evolution. Extensive germplasm resources, largely a byproduct of their economic importance, together with growing collections of defined mutants, provide foundations for a host of post-genomic studies to shed more light on the relationship between sequence and function in this important group. Using the rapidly growing capabilities of several informatics resources, genomic data from model cereals are likely to be leveraged tremendously in the study and improvement of a wide range of crop plants that sustain much of the world's population, including many which still lack primary genomic resources.
The cultivated cereals, members of the Poaceae family of the angiosperms, provide about half of the calories consumed by humans and a growing share of biofuel. Together with their economic importance, the Poaceae are an attractive group for comparative genomics because they include many important crops with diverse native distributions and at least 35-fold variation in genome size (e.g., rice = 420 Mb; wheat = ∼15,000 Mb). The independent domestication of rice in both Africa and Asia, sorghum in Africa, maize in America, and wheat in the Near East has provided an excellent study system in which to explore the genetic complexity of adapting plants to human use (for example, see Paterson et al. 1995; Paterson 2002).
Recent efforts to characterize Poaceae genomes better are reflected in their expansion from 1% to about 6% of the DNA sequence resources in GenBank (Paterson et al. 2003). This is exemplified by the nearly finished sequencing of each of two Oryza subspecies (see below), supplemented by exploratory genome-wide efforts in maize (Whitelaw et al. 2003) and sorghum (Bedell et al. 2005), and large EST and STS-based DNA marker collections for many others.
Collectively, genomic resources for diverse Poaceae promise new insights into molecular evolution, botanical diversity, and agricultural productivity. The power of any family or clade as a system to answer fundamental questions depends largely on the number of whole-genome sequences available, the exact branch lengths and positions of these data sets in the phylogenetic tree, and the positions of whole-genome duplications in the tree. For efficient comparison, orthologous genes or regions cannot be too closely or too distantly related; grass divergence events happened at useful times. Even if the grass family were represented solely by the whole genomes of rice, sorghum, and maize, their relationship just happens to confer tremendous analytic power to unravel much of the evolutionary history of both entire genomes (Fig. 1A,B) and individual genes (Fig. 2) in this important family. In grasses is the happy union of economic and scientific interests.
Rice
Rice is considered a model cereal crop because it has a relatively small genome size as compared with other cereals, a vast germplasm collection, an enormous repertoire of molecular genetic resources, and an efficient transformation system. The scientific value of rice is further enhanced with the elucidation of the genome sequence of the two major subspecies of cultivated rice, Oryza sativa ssp. japonica and ssp. indica. The sequence of the japonica cultivar Nipponbare was recently completed by a consortium of 10 countries, which comprised the International Rice Genome Sequencing Project (IRGSP), and represents a map-based finished sequence of the entire genome obtained using the hierarchical clone-by-clone sequencing strategy (Sasaki and Burr 2000). The sequence of the indica cultivar was derived by a whole-genome shotgun sequencing approach (Yu et al. 2002, 2005). These genome sequences are invaluable resources not only in understanding the structure and function of the rice plant itself but also in deciphering the organization of other cereal genomes, which share an appreciable degree of synteny with rice (e.g., Paterson et al. 2004; Devos 2005).
Genome organization and sequence
A total of 370 Mb of finished sequence of PAC and BAC clones from japonica rice, including virtually all of the euchromatic regions, revealed several characteristic features of the rice genome. A total of 57,000 protein-encoding sequences were inferred by computational gene predictions from all finished sequences (Yu et al. 2005). This is definitely an overestimate because we know that a large fraction of these (13% in chr. 1 and 18% in chr. 10) are various kinds of transposable elements (TEs) (Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). Exclusion of all TE-related predicted genes would reduce the estimated gene number to about 43,000, implying a much lower gene density than the 4.5/kb value for Arabidopsis (Arabidopsis Genome Initiative 2000).
The map-based sequence also provides accurate positional information of genome components characterized by the presence of repeat or exogenous sequences. Widespread gene transfer from the organelles is suggested by the presence in the nuclear genome of many chloroplast and mitochondrial DNA fragments, including some nearly intact copies. The sequence of the centromeres revealed clusters of highly repetitive CentO satellite DNA located within the functional domain and flanked by centromere-specific retrotransposons.
A draft sequence of the indica subspecies representing 4.2× genome coverage and comprising 103,044 scaffolds corresponding to 466 Mb (Yu et al. 2002) was recently increased to 6.28× coverage and further revised using the Syngenta Nipponbare whole-genome shotgun sequence data (Goff et al. 2002) resulting in a total genome assembly of 466.3 Mb (Yu et al. 2005). A total of 19,079 nonredundant Nipponbare full-length cDNAs (97.7%) were localized and 49,088 genes were predicted. The discrepancies in gene numbers between japonica and indica genomes illustrate the need for more uniform and accurate methods of gene identification. In the japonica genome, this concern is being addressed through manual curation of annotation using full-length cDNA information.
Gene functional assignments and resources
In addition to the assembled genomic sequences, public databases currently contain 386,487 rice ESTs (dbEST release 031105). A collection of more than 32,000 nonredundant rice full-length cDNA sequences is also publicly available (Kikuchi et al. 2003). These resources together with the accurate map-based genome sequence will be indispensable for further functional characterization of the rice genome. To address the huge task of determining the function of all predicted genes in rice, the International Rice Functional Genomics Consortium has been established using much the same organizational model as the IRGSP (Hirochika et al. 2004). The functional genomics resources currently available and which can be used for both forward and reversed genetics methods include insertion mutant lines generated using Tos17, T-DNA, and Ac/Ds elements, as well as populations of deletion mutants.
The map-based sequence has proven especially useful for the identification of genes underlying diverse agronomic traits such as flowering time, plant architecture and development, fertility restoration, and disease resistance. Many of these traits are governed by multiple genetic loci or QTLs. The genome sequence facilitates the development of DNA markers for this analysis. At least three genomewide SNP discovery studies have been conducted, with marked differences in numbers and rates of inferred SNPs due to the use of different target sequences (low-copy vs. total genomic vs. BAC-end) and different approaches to filter paralogs and artifactual SNPs from the respective data sets. Comparison of low-copy DNA across the entire japonica and indica sequences using short sequence alignments and stringent filtering criteria resulted in inference of 408,898 (Feltus et al. 2004) SNPs or about 1 per kb across the genome (but 1.7 per kb in the low-copy DNA that was screened), of which about 80% could be empirically verified in a sample. Analysis of total genomic DNA (Zhao et al. 2004) suggested ∼5 million SNPs using longer alignments but looser stringencies to filter paralogs and false positives. Comparison of the japonica sequence with the end sequences of BAC clones from another indica cultivar, Kasalath, revealed a SNP frequency of 0.71% (Katagiri et al. 2004), intermediate between the other two studies.
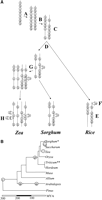
Unraveling the history of cereal gene and genome evolution. (A) A whole-genome duplication A and associated divergence and/or loss B of some members of duplicated gene pairs, determined the gene set that was inherited by all cereals C. After divergence of the cereals from this common ancestor D, continuing gene loss (E: note that locus e has been preserved on one homoeolog in the indicated lineage and the other homoeolog in the alternate lineage, thus changing its linkage relationship to flanking genes) together with the effects of other rearrangement mechanisms such as transposition F, led to incongruities in gene arrangement of modern cereal crops. Additional whole-genome duplications in some lineages such as maize, sugarcane, and wheat G, accompanied by continuing activity of transposition mechanisms H, resulted in further differentiation of the modern gene repertoire and order from that of the ancestral cereal order. Finished sequences of genomes representing the major taxonomic groups within the cereals will permit unraveling of the nature and timing of many of the events that account for such differences, as well as inference of the general organization of the genome of their common ancestor. (B) Phylogenetic relationships among selected Poaceae lineages. Current thinking on the approximate relationships among the major lineages discussed are illustrated. (*) The Sorghum genus includes a recently formed polyploid, Johnson grass (S. halepense). (**) The Triticum lineage includes several recently formed polyploids, most notably tetraploid T. durum (durum wheat) and hexaploid T. aestivum (bread wheat).
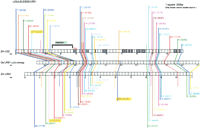
Two independent BLAST comparisons plotted coordinately: maize gene lg2 (AY180106 from a BAC) with its rice ortholog and maize gene lrs1 (AY180107 from a BAC) with the same rice ortholog (RGP PAC AP003287). The purple alignment line of lg2CNS17 represents how sequences near 5′ exons align. Exons were identified in genomic DNA for all three genes using the complete cDNA of lg2-mRNA (AF036949) and then masked. Bl2seq conditions were modified from those of Kaplinsky et al. (2002b). The BLAST result is represented by the solid, multicolored lines connecting the maize lg2 (Zm LG2) and rice lrs1 (Os LRS1) gene diagrams. The identity match is indicated parenthetically. Each color connects CNSs that are essentially the same. A broken line reflects a maize lg2-lrs1 CNS retention, but just below the 15/15 cutoff. Yellow highlighting denotes those rare CNSs that are fractionated. Fractionation at the DNA level could, hypothetically, explain subfunctionalization at the phenotypic level. An insertion into lg2 promoter is noted. Figure was reproduced with permission from the Genetics Society of America © 2004, from Langham et al. (2004).
Mapping populations such as recombinant inbred lines (RILs), backcross inbred lines (BILs), doubled haploid lines (DHLs), and chromosome segment substitution lines have been developed for rice to facilitate identification of target genes (Yano and Sasaki 1997).
Sorghum
The #5 grain crop worldwide based on tonnage (after wheat, rice, maize, and barley; see http://www.fao.org), sorghum is unusually tolerant of low input levels, an essential trait for agriculture in areas that receive little rainfall, such as Northeast Africa and the U.S. Southern Plains. The Sorghum genus also includes one of the world's most noxious weeds. Many features that make “Johnson grass” (S. halepense) such a troublesome weed are actually desirable in forage, turf, and biomass crops, adding further value to enhanced knowledge of the Sorghum genus.
As a model for the tropical grasses, sorghum is a logical complement to Oryza (rice: Fig. 1B). Sorghum is representative of tropical grasses in that it has C4 photosynthesis, biochemical and morphological specializations that improve net carbon assimilation at high temperatures. By contrast, rice uses C3 photosynthesis, more typical of temperate grasses. Sorghum and maize shared a common ancestor about 12 million years ago (Mya)— however, the sorghum genome is much smaller (∼736 Mbp), due to both recent polyploidization and repetitive DNA propagation in maize (see below). Sorghum is an even closer relative of sugarcane, arguably the most important biomass/biofuel crop worldwide (http://www.fas.usda.gov). However, sugarcane has out-done maize, undergoing at least two whole-genome duplications in the ∼5 Myr since divergence from sorghum (Ming et al. 1998), resulting in a genome larger than that of human and with 4-fold or greater redundancy of most genes. While this article was in review, the U.S. Department of Energy Joint Genome Institute Community Sequencing Program announced its intention to perform 8× whole-genome shotgun coverage of the Sorghum bicolor genome, for public release.
Genome organization and sequence
The most detailed sorghum sequence-tagged site (STS)-based map is from a cross between Sorghum bicolor (SB) and S. propinquum (SP), comprising 2512 restriction fragment length polymorphism loci that span 1059.2 cM (Bowers et al. 2003). A total of 865 heterologous probes link the sorghum map to those of Saccharum (sugarcane: Ming et al. 1998), Zea (maize: Bowers et al. 2003), Oryza (rice: Paterson et al. 1995, 2004), Pennisetum (millet, buffelgrass: Jessup et al. 2003), the Triticeae (wheat, barley, oat, rye), Panicum (switchgrass: Missaoui et al. 2005), and Cynodon (bermudagrass: C. Bethel, E. Sciara, J. Estill, W. Hanna, and A.H. Paterson, in prep.).
Sorghum was the first plant for which a BAC library was reported (Woo et al. 1994). Physical maps of both SB and SP have been constructed and genetically anchored by hybridization to the mapped STS loci (Bowers et al. 2005). In a recent assembly (available at http://cggc.agtec.uga.edu), 40,957 SP BAC fingerprints (7× genome coverage) yielded 1541 contigs (averaging 24.4 BACs and 12.5 hybridization loci), while 69,545 SB BACs (11× coverage) yielded 1869 contigs (35.6 BACs, 10.1 hybridization loci). These assemblies exclude 100% of “Q” (questionable) clones. About 200 contigs exceed 1 million bp, and another ∼500 are >500 kb. The contigs appear to cover >90% of the sorghum genome.
Sorghum was the first organism for which Cot-based reduced-representation sequencing approaches were reported (Peterson et al. 2002a), yielding extensive coverage of its repetitive DNA sequence and a promising means to eventually obtain most of its low-copy DNA (Peterson et al. 2002b). A complementary resource providing about 1× coverage of the hypomethylated DNA of sorghum has been reported recently (Bedell et al. 2005).
Gene functional assignments and resources
As of this writing, about 170,000 sorghum ESTs were available in GenBank, many from a carefully structured experiment that produced ∼5000 ESTs from each of 10 libraries representing diverse tissues and developmental stages (Cordonnier-Pratt et al. 2005). Nearly 300,000 ESTs for sugarcane are also available (Vettore et al. 2003).
While its minimal level of gene duplication makes sorghum, like rice, an attractive system for many approaches to determining gene function, this opportunity has been underexploited. Past efforts by the U.S. Department of Agriculture Agricultural Research Service have resulted in a collection of several hundred mutants (C. Franks, pers. comm.), but only a small subset have been studied formally. Identification of an active transposable element in sorghum (Chopra et al. 1999) has laid the foundation for insertional mutagenesis in sorghum.
Maize
Maize (n = 10) is a recent domesticate of the tropical grass teosinte (Doebley 2004). The most recent maize whole-genome duplication happened approximately 12 Mya (Gaut and Doebley 1997). Sorghum (n = 10), with its clearly diploid genome (Chittenden et al. 1994; Moore et al. 1995), differentiated from a maize ancestor just before this tetraploidy (Swigonova et al. 2004). Over the 12 Myr since tetraploidy, the maize genome “diploidized” by deleting most of the duplicated centromere regions and also deleting or tolerating degeneration of one member of most of its paired gene sets, sometimes fragmenting ancestral gene orders across multiple chromosomes and obscuring similarities in gene order that existed among its ancestors (Fig. 1A). The results of fractionating a tetraploid back toward a diploid can be complicated (e.g., Song and Messing 2003; Brunner et al. 2005). Comparisons of sorghum and maize chromosomal details should facilitate condensing maize back onto the tribal (Andropogonae) and eventually family (grass) ancestral genome. While all flowering plants are paleopolyploids, maize has largely been restored to diploidy.
Few if any plants rival the contributions of maize as a Mendelian genetic model during the 20th century (MaizeGDB [http://maizegdb.org] serves as a clearinghouse for maize genetic information). Today, maize continues to be a leading botanical model system for analysis in several areas, including transposons (Lisch 2002; Kolkman et al. 2005), meiosis where visualization of chromosomes is important (e.g., Harper et al. 2004), and the limits of fine mapping of phenotypes (Wright et al. 2005). Not all maize inbred lines carry identical gene contents (Fu and Dooner 2002); heterosis could well be a result. Transposons have been implicated as a mechanism generating such rapid intraspecies movement and diversity (Lai et al. 2004; H. Dooner, pers. comm.).
Genome organization and sequence
The 21st century finds maize in the process of being sequenced. With an estimated 2300-2600 Mb of chromosomal DNA (6× rice and 20× Arabidopsis), of which at least 60% is retrotransposon, the maize genome has initially been “filtered” to enhance for unmethylated (Rabinowicz et al. 1999) or low-repeat (Peterson et al. 2002a; Yuan et al. 2003) sequence—before shotgun sequencing (Whitelaw et al. 2003). The up-to-date result, best seen at the appropriate TIGR Web page (http://maize.tigr.org/), is about 1 million reads of about 700 bp each from inbred line B73. Additional reads, from other genotypes, are also available in GenBank. Three independent groups are assembling these sequences into contigs, defining what has come to be called “gene space.” Without doubt, this filtration method could miss particular genes, genes that will only be found in unbiased genomic sequence. Even so, the vast majority of nonrepetitive DNA in maize is represented by the current genome survey sequences. At press time, the U.S. National Science Foundation is considering proposals to “finish” the maize genome.
Gene functional assignments and resources
Maize genetics enjoys a host of both public and private resources. MaizeGDB holds the current maps, nomenclatures, gene content data, descriptions and photographs of mutant phenotypes, and references, and lists the positions of thousands of BACs and mapping markers, markers that are often also useful in grasses other than maize. The 407,423 maize ESTs have been used by both TIGR and MaizeGDB to form gene lists; TIGR finds about 58,500 gene-like units in maize. Based on these or similar gene sequences, two projects—Pioneer Hi-Bred's TUSC (Trait Utility System for Corn) and Maize Targeted Mutagenesis (MTM)—permit users to order seeds homozygous for Mu transposon insertions potentially knocking out any gene that has been sequenced. Two public projects generated ears segregating for mutants that are likely caused by Mu transposon insertion. These phenotypes are often photographed or described at the MTM or MaizeGDB Web sites; phenotypes may also be screened and seed may be ordered from the Maize Genetics Cooperation Stock Center's Web site (http://w3.aces.uiuc.edu/maize-coop/). Transposon Mu has been a particularly useful mutagen in maize (Lisch 2002) and allows for subsequent tagging and cloning by a variety of methods (Brutnell 2002). Forward genetics on essential genes can be challenging: A maize “TILLING” service (Till et al. 2004) delivers point mutants given gene sequence (http://genome.purdue.edu/maizetilling). Adding genes to maize is possible but time-consuming; alternative public projects can help add a user's gene to maize within a T-DNA (http://www.biotech.aistate.edu/service_facilities/plant_transformation.html) or by biolistics (http://www.psu.missouri.edu/muptcf/service/customized_service.htm). The maize ESTs, or derived synthetic DNAs, have been used to make hybridization microarrays. Transcript profiling experiments may be found at MaizeGDB and links therein.
Cross-taxon messages
For several reasons, the cereals are a particularly promising family in which to answer fundamental questions about many aspects of plant genome evolution. Their ∼35-fold variation in genome size (c-value), while sharing a largely common set of genes, invites questions into evolution of genome size and structure. The fact that they all share a whole-genome duplication event that occurred “shortly” (ca. 20 Myr) before their divergence invites inquiry into the range of fates of ancient homeologs in reproductively isolated lineages. Several recent duplications (sugarcane, maize) or polyploidizations (wheat, Johnson grass) provide potential systems in which to explore repeating patterns in the fates of genes and gene families, as exemplified in Figure 1. Correspondence in the chromosomal locations of genes controlling many traits, for example, key features of domestication (Paterson et al. 1995) and weediness (Hu et al. 2003), highlights the many leveraging opportunities for information about model cereals. Finally, their economic importance makes a strong case for sequencing of the genomes of additional cereals and provides a large community to help translate such investments into agricultural benefits. We elaborate on a few promising early opportunities below.
Comparative gene fates
A natural first question posed of the second genome sequenced in a taxonomic group is whether taxon-specific genes can be discerned. That is, will the analysis of well-annotated versions of their completed genomes reveal genes that are present in rice and maize (for example) but absent (or at least unrecognizable) in Arabidopsis, Populus, Medicago, or other flowering plants? At present, approximately 71% of predicted rice proteins have a homolog in Arabidopsis whereas a total of 2859 genes appear to be unique to rice and other cereals. These numbers must be considered preliminary estimates, as inferences of the existence of taxon-specific genes are complicated by differences in sequence annotation methods, evolutionary rates of entire genes and specific motifs, chronic problems with sequencing of heterochromatic regions, and other obstacles. Nonetheless, the finding that specific transposable element families in rice may occasionally “stitch together” new genes from parts of existing ones (Jiang et al. 2004) makes the cereals an especially interesting group to explore for such events.
While positional correspondence of many genes across the cereals is well known, functional correspondence among the vast majority of these genes remains a hypothesis to be tested. All cereals are ancient polyploids, most recently as a result of a duplication event thought to have occurred perhaps 20 Myr prior to their divergence from common ancestors. This 20-million-year lag appears to contraindicate the possibility that polyploidization contributed directly to species divergence (e.g., Lynch and Force 2000) in the cereals but opens the door to the possibility of extensive “subfunctionalization” (Lynch and Conery 2000), with pairs of ancient homoeologs each undergoing compensatory mutations that result in subdivision of the ancestral gene's function. Further, post-polyploidization gene loss appears to have still been happening 20 million years later, in that loss of different members of homoeologous sets accounts for a sizable portion of apparent deviations from gene order conservation in rice and sorghum (Paterson et al. 2004). If subfunctionalization or even neofunctionalization (the acquisition of entirely new function) had preceded loss of different members of homoeologous sets in the two lineages, then the phenotypic consequences could be substantial. A host of genomic data will contribute to investigation of the degree to which functional correspondence parallels positional correspondence of individual genes, particularly including genomewide expression profiling, and saturation mutagenesis, providing empirical data with which to develop and test hypotheses about readily observable differences among cereals in sequence and structure of specific genes and their flanking regions.
Conserved noncoding sequences: Approaching gene regulation computationally
All but a few of the most basal grasses radiated into subfamilies about 50 Mya (Kellogg 2001). An estimated 20 Myr prior to this radiation, the grass lineage had a whole-genome duplication, with about 20% of genes being retained as pairs within modern rice and many other modern grasses considered to be diploids (Paterson et al. 2003, 2004, 2005; Vandepoele et al. 2003; Wang et al. 2005; Yu et al. 2005). By multiplying the published rate of neutral base-pair substitution in grasses by the divergence time, it was calculated that a 15/15-bp exact positional match between orthologous genes in maize versus rice (or, coincidentally, man vs. mouse) would occur without selection only four times in a million; pairwise noncoding alignments with e-values equal to or more significant than 15/15 within a gene space define a grass conserved noncoding sequence (CNS) (Kaplinsky et al. 2002). So, alignments between retained duplicates within all grasses and comparisons between orthologs representing different grass subfamilies have diverged for enough time to insure CNS function but have not diverged so much that alignments are lost. As with mammals, the number of sequenced or in-progress grass genomes and their exact placement in the phylogenetic tree provide a powerful experimental system for studying the relationships between CNSs, gene regulation, and phenotype (Lockton and Gaut 2005). Grasses have far fewer and much smaller CNSs than do mammals, a possible indication of relative developmental simplicity (Inada et al. 2003). This relative simplicity has experimental advantages. For one case, CNS function involves a CNS-rich 1.5-Kb region of intron within the knotted Class I homeobox gene in grasses (Inada et al. 2003); mutant phenotypes in maize indicate that transposon insertions in this region can prevent a negative function keeping this gene “off” in wild-type leaves (Greene et al. 1994), which is the sort of regulatory switch often accomplished at the chromatin level. See Lockton and Gaut (2005) for a review of plant CNS research and on the power of having usefully placed outgroup genes for CNS research. A graphic example of maize-rice CNS alignments and possible subfunctionalization is illustrated in Figure 2. CNSs found in maize-rice comparisons have been shown to work well as specific PCR primer sites. These sites are conserved in grasses representing the breadth of grass diversity (Kaplinsky et al. 2002). Thus, CNS research between any two usefully diverged grasses delivers pan-grass PCR mapping and sequence-extraction tools.
Heterochromatin and its importance
In large repeat-rich plant genomes, sequencing plans naturally prioritize relatively gene-rich euchromatin, both because of its information yield and relative ease of assembly. It is important, however, that heterochromatic (particularly pericentromeric) regions be sequenced to the degree possible at least in a few models to permit exploration of their unique features. It is a widespread observation that recombination (in the sense of reciprocal chromosomal exchange) is rare in pericentromeric regions. Suppression of recombination, together with a high tolerance for repetitive DNA, may create the sort of genomic environment that would favor the evolution of “co-adapted gene complexes,” which are predicted to be favored by domestication (D'Ennequin et al. 1999) and have been observed in several cases (Paterson 2002). Identification and characterization of such complexes would be very important in a wide range of contexts. Early progress toward this goal has been accomplished for individual centromeres (Nagaki et al. 2004).
Needs and opportunities
The availability of so many economically important taxa within the Poaceae family has made it a logical focus of monocot genomics. Finishing the maize genome, together with the sequencing of sorghum and at least one member of the Triticeae group including wheat and barley (a very important group that was not otherwise addressed in this manuscript), is a natural priority. A good case can also be made for several other family members— for example, foxtail millet (Setaria spp.) has a small genome and represents the sister tribe to the one containing maize-sorghum-sugarcane (thus, it is an appropriate outgroup for studies of these tropical grasses). It also contains some of the toughest, weediest species and landraces in the plant kingdom, offering new genes to help confer on our major crops the ability to remain productive under desertification.
It would be of direct value to Poaceae genomics to have extensive resources (preferably a complete genome) for a closely related taxon that could provide an “outgroup” for phylogenetic triangulation of events at a wide range of levels ranging from gain/loss of individual genes and parts of genes to chromosomal-level rearrangements. Within the two orders closest to the monocot suborder Poales, the Zingiberales includes banana (Musa spp.), a genome with 2n = 18 and 1C = 600 Mbp, which is of major global importance in terms of food and income security to millions of smallholder farmers throughout the developing countries of the tropics and subtropics. Further, the Commelinales includes pineapple, Ananas comosus L., a diploid, self-incompatible, with 2n = 50, and 1C = 526 Mbp (Arumuganathan and Earle 1991). Besides being an important fruit crop, pineapple represents still another strategy for carbon fixation (CAM). Its abnormally high somatic mutation rate (Collins 1936, 1960) suggests that it, like sorghum and rice, may prove to be a facile system for modern approaches to relate gene sequence to function.
While most Poaceae genes are now identifiable from current or pending genomic sequences, there remain many gaps in knowledge of the patterns of intragenic variation across the major branches of the family. Particularly glaring is the lack of knowledge of the Bambusoids (bamboos), for which only 295 sequences are found in GenBank as of this writing; the Chloridoids including major turfgrasses such as Bermuda (Cynodon) and zoysia (Zoysia) and orphan crops such as tef (Eragrostis tef), with 5367 sequences as of this writing; and Arundinoids, including the reeds, with only 178 sequences. One can also make good cases for more information about several lower-level taxonomic groups within the better-studied branches of the Poaceae.
Much of the value of whole-genome sequences in the cereals is likely to be realized by analysis of levels and patterns of allelic diversity in cultivars and their wild relatives. Germplasm collections are the underpinning of crop improvement—however, the 300,000+ accessions in global cereal genetic resources collections are woefully underexplored. Association genetics approaches (Thornsberry et al. 2001) are a promising tactic by which to begin to realize the value that lies latent in these resources; however strategies will have to accommodate species-specific features of the structuring of diversity. For example, linkage disequilibrium (LD) decays rapidly over a few hundred base pairs in maize (Remington et al. 2001), but in sorghum average LD (r2) in six unlinked genomic regions only falls below 0.2 for pairs of sites >15 kb apart (Hamblin et al. 2005). Thus, where gene-by-gene approaches may be necessary in maize, LD-based association genetics appears more promising in sorghum.
Realizing the opportunities: Database resources
Realization of the cross-taxon messages that lie latent in the Poaceae will require still more attention to integrative computational resources. Increasing complexity of data and sophistication of user queries make the appropriate content and organization of such databases a moving target, and in addition complementary resources are needed for additional taxa. Gramene (Ware et al. 2002; http://www.gramene.org) represents a valuable early effort toward a pan-Poaceae genomic database from a rice-centric perspective, providing a platform for comparison of rice mapping and sequence information with corresponding data in other cereal crops.
The rice genome sequencing effort has been accompanied by generation of several databases. The Rice Genome Automated Annotation Sytem (RiceGAAS: http://ricegaas.dna.affrc.go.jp) is both an annotation tool and a database of the annotation of all rice genome sequences ranging from 10 kb to 1 Mb submitted to GenBank. Manually curated annotation of the Nipponbare genome sequence can be accessed through the Rice Annotation Database (RAD: http://rad.dna.affrc.go.jp) and the Integrated Rice Genome Explorer (INE: http://rgp.dna.affrc.go.jp/giot/INE.html). Furthermore, INE integrates the genome sequence information with the genetic map, physical map, and transcript map of rice. The TIGR Rice Genome Annotation Database http://www.tigr.org/tdb/e2k1/osa1/) also features the sequence and annotation data for the rice genome. Finally, Oryzabase (http://www.shigen.nig.ac.jp/rice/oryzabase/top/top.jsp) provides information on classical rice genetics as well as current advances in rice genomics.
While there are many online resources supporting research on maize, most of them may be accessed through the “links” button at the top of the MaizeGDB homepage. MaizeGDB is “information central” for maize research, both basic and commodity driven. The Maize Page also carefully links maize resources (http://maize.agron.iastate.edu). For example, while the INRA Maize Genomic Database (http://moulon.moulon.inra.fr/imgd)—a must visit for all QTL studies—was not mentioned in the paragraph on maize above, its link may be found readily.
For sorghum, an online resource (http://cggc.agtec.uga.edu) provides access to sorghum EST, genetic/physical map, and polymorphism data via Web interfaces and bulk downloads. Underlying this resource is an Oracle relational database containing tables for data storage, display parameters, and links to external Web sites as well as database views to implement on-line statistical calculations, with flexibility for a wide range of query options. Examples of Web-accessible displays are the IntegratedMap display that provides a graphic view of the most detailed sorghum STS-based genetic map along with mapped polymorphisms and anchored BAC contig data, the OxfordGrid display for comparative genetic map views, and the ESTminer display that shows contigs and clusters as color-coded tree nodes with expandable cDNA library statistics. The Web displays are compatible with a wide range of browsers and operating systems. Additional complementary Web-based resources focus on functional genomics of the transcriptome (http://fungen.botany.uga.edu) and genomics of the unique abiotic stress responses of sorghum (http://sorgblast2.tamu.edu). Finally, online resources from SUCEST (http://sucest.lbi.dcc.unicamp.br/en/), an extensive EST project in closely related sugarcane (Vettore et al. 2003), are also often of value for sorghum genomics.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3725905.
-
↵4 Corresponding author. E-mail paterson{at}plantbio.uga.edu; fax (706) 583-0160.
- Cold Spring Harbor Laboratory Press








