
Simple, robust methods for high-throughput nanoliter-scale DNA sequencing
Abstract
We have developed high-throughput DNA sequencing methods that generate high quality data from reactions as small as 400 nL, providing an approximate order of magnitude reduction in reagent use relative to standard protocols. Sequencing of clones from plasmid, fosmid, and BAC libraries yielded read lengths (PHRED20 bases) of 765 ± 172 (n = 10,272), 621 ± 201 (n = 1824), and 647 ± 189 (n = 568), respectively. Implementation of these procedures at high-throughput genome centers could have a substantial impact on the amount of data that can be generated per unit cost.
Between February 2004 and February 2005 the NCBI Trace Archive (http://www.ncbi.nlm.nih.gov/Traces) received 287,319,810 sequencing read electropherograms from various high-throughput DNA sequencing projects. A nominal cost of $1 per read suggests that in its most strict definition, high-throughput DNA sequencing is presently at least a several hundred million dollar per year industry. Large-scale genomics efforts, particularly whole genome sequencing and polymorphism detection sequencing, continue to be cost-limited. Current efforts to increase data production per unit cost have focused on either (1) new and potentially revolutionary methods such as sequencing by synthesis (Brenner et al. 2000), polymerase colony sequencing (Mitra et al. 2003), and single molecule sequencing (Braslavsky et al. 2003; Levene et al. 2003); or (2) on evolutionary approaches that strive for volume reduction within the paradigm of Sanger sequencing (Sanger et al. 1977), four-color fluorescence (Smith et al. 1985, 1986), and capillary electrophoresis (Dovichi 1997). Here we focus on the latter approach. Typically, high-throughput genome centers use sequencing reaction volumes of several microliters, which yield quantities of products far in excess of what is required to generate high-quality data by capillary electrophoresis. The limiting factors for volume reduction have been reaction vessels (microtiter plates) that allow sample loss by seal leakage and condensation on unwetted inner well surfaces (see Supplemental material) and rigid adherence of laboratory liquid-dispensing robotics to these established microtiter plate formats.
Here we describe how testing and implementation of (1) a novel nanoliter-scale reaction vessel configuration, (2) submicroliter positive pressure microsolenoid-based liquid-dispensing robotics, and (3) optimized sequencing reaction chemistry, thermocycling conditions, and capillary electrophoresis injection parameters have allowed us to generate high-throughput sequence data of equal or greater quality to standard methods, with substantial reduction in reagent consumption. All materials we describe are commercially available, such that with appropriate attention to detail the process we describe can be easily implemented at other facilities.
Results and Discussion
Validation tests were performed on 16 different libraries constructed using a variety of common low-, medium-, and high-copy number vectors (Table 1).
Plasmid, BAC, and fosmid DNA libraries sequenced in a 400-nL reaction containing 31.25 nL of Big Dye terminators (V3.1, Applied Biosystems) as compared to standard 4000-nL reactions containing 540 nL of Big Dye terminators
For plasmid libraries, mean PHRED20 read lengths in excess of 750 bp were achieved (Fig. 1A). For library 10790, which is a mock library in which every well contains the same human cDNA clone, the average read length approached 900 bp, and the longest read was 972 bp (Supplemental Fig. 1). These read lengths were comparable to those achieved with our standard DNA sequence production pipeline that uses 540 nL Big Dye Terminator Premix V3.1 (Applied Biosystems) (Table 1), and comparable to those typically submitted to the NCBI trace archive by other high-throughput centers. For all templates that were sequenced in this study, excluding the ∼4% failed cultures, the average sequencing success rate (samples with a PHRED20 read length of at least 100 bp) was 97%. Although all data in the present study are single-end reads, when applied to paired-end sequencing this method would be expected to give a successful paired-end rate of 0.97 × 0.97, or 94%. After sequencing these libraries using 31.25 nL of Big Dye terminators per well and obtaining adequate read length and signal strength (Table 1), we investigated the absolute lower limit for Big Dye terminator consumption. Ninety-six identical clones from library 10790 were sequenced using either 15.63 nL, 7.81 nL, or 3.13 nL of Big Dye terminators in a 400-nL total reaction that contained 20 nL of 15X reaction buffer; 1 nL, 0.5 nL, or 0.2 nL, respectively, of 100 μM -21 M13 forward primer; and an appropriate volume of Ultrapure water (Invitrogen). Mean PHRED20 read lengths obtained were 871 ± 97, 684 ± 92, and 0 bases, with maximum PHRED20 read lengths of 887, 703, and 0 bases, respectively (Fig. 1B), suggesting that the limiting volume of dye terminators in this system lies between 3.13 and 7.81 nL.
We expect that given the simplicity of the platform we describe here, and its foundation firmly within the time-tested Sanger sequencing paradigm, it will be easily implemented by any center engaged in moderate- to high-throughput DNA sequencing. The chemistry is robust for all vector types (plasmids, fosmids, BACs) typically utilized for high-throughput sequencing. While reagent, equipment, and labor costs will vary among high-throughput sequencing platforms at different institutions, implementation of these methods at our center reduces the cost of sequencing reactions by ∼90%, relative to the cost of our established 4-μL reactions (Supplemental Fig. 3). We expect that cost savings realized from volume reduction can very rapidly offset the cost of the robotics and plasticware required for the process.
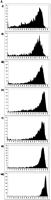
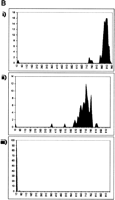
(A) Distribution of read lengths (PHRED20 base count) for each library described in Table 1, sequenced with a 400-nL reaction that contained 31.3 nL of Big Dye terminators (v3.1, Applied Biosystems). (i) CA001, CD001, CE001, CL001: Fosmid end reads; (ii) GA000: BAC end reads; (iii) CN23E: medium copy-number plasmid whole genome shotgun reads; (iv) LL005-LL0017: high copy-number plasmid 5′ EST reads; (v) TX060, TX067: high copy-number plasmid transposon-mediated shotgun reads; (vi) S1881: high copy-number plasmid SAGE library reads; (vii) 10790: high copy-number plasmid 5′ EST reads from 2304 identical clones. (B) Distribution of read lengths (PHRED20 base count) for 96 identical clones from library 10790, sequenced with a 400-nL reaction that contained (i) 15.63 nL, (ii) 7.81 nL, or (iii) 3.13 nL of Big Dye terminators.
Methods
A flow diagram summarizing the basic process is presented in Supplemental Figure 2. Template DNA from multiple libraries constructed with different vector types (Table 1) was prepared as previously described (Yang et al. 2005). Briefly, for plasmid clones, 60 μL of 2xYT liquid culture was grown for 18 h with shaking (350 rpm) in 384-deep well diamond plates (Axygen) covered with AirPore tape (Qiagen). Only sample wells that failed to show any cell growth (4%) were removed from analysis. To extract DNA, 60 μL of lysis buffer (Qiagen) was added directly to the overnight culture. After 5 min of lysis, 60 μL of neutralization buffer (Qiagen) was added. Plates were tape sealed (Edge Biosystems clear tape) and mixed by vortexing on a high-power multi-plate vortexer (VWR, model VX-2500) at maximum speed for 2 min prior to centrifugation at 4250g for 25 min. One hundred twenty microliters of cleared lysate was transferred from culture blocks into 240-μL 384-deep well diamond plates containing 90 μL of 100% isopropanol per well, mixed by inversion, and centrifuged at 2830g for 15 min. Isopropanol was decanted, and the DNA pellet was washed with 50 μL of 80% ethanol and then air-dried. DNA pellets were resuspended in 10 mM Tris-HCl pH 8 containing 10 μg/mL RNase A (Qiagen). BAC DNA was prepared using a similar automated alkaline lysis procedure in 96-well format, as previously described (Schein et al. 2004). These are generally very crude DNA preparations, as no organic solvents, paramagnetic particles, membranes, or filters are used in the process. DNA was quantified using pico green against a standard curve generated using known amounts of phage lambda DNA.
After careful consideration (see Supplemental material) we determined that the most practical method for delivering DNA template to a submicroliter reaction was to transfer a relatively large volume of dilute template using standard laboratory robotics, desiccate, and then resuspend in an appropriately small volume (200-400 nL) of sequencing reaction master mix. Using a Biomek FX (Beckman-Coulter), plasmid DNA was diluted 10-fold in Ultrapure water (Invitrogen), and 2 μL (∼15-55 ng) was transferred to 384-well PCR cycle plates (ABgene), then completely desiccated in a drying oven for 10 min at 95°C. We find that it is important to restrict the volume of the initial transfer to ≤2 μL, as DNA that adheres to the inner well surface during desiccation will be unavailable for the sequencing reaction. High-throughput sequencing centers that serially process a large number of plates may wish to dry the plates overnight at room temperature in a laminar flow hood, as we have found this to work just as well as a drying oven.
Subsequent to template transfer, 400 nL of sequencing reaction mix containing 31.25 nL Big Dye Terminator Premix V3.1 (Applied Biosystems), 40 nL of a custom formulation of reaction buffer (25X Reaction Buffer [2M Trizma Base, 50 mM MgCl2-6 H2O] combined with an equal volume of 5X Big Dye Terminator Reaction Buffer V3.1 [Applied Biosystems]), 2 nL of 100 μM primer (Invitrogen), and 326.75 nL Ultrapure Water (Invitrogen) were added to each well using the Aurora Discoveries Flying Reagent Dispenser. This instrument was developed for high-throughput screening in the pharmaceutical industry and has not previously been applied to high-throughput sequencing applications (Supplemental material). Plates were sealed with SPRI Plug Low Volume lids (Agencourt Bioscience) to reduce residual air space in the wells and thereby reduce sample loss by evaporation. Again, the principal limiting factor for volume reduction of cycle sequencing has been loss of fluid from the sample due to elevated temperatures and rapid temperature changes (please refer to Supplemental Online Material for theoretical consideration of factors contributing to fluid loss and measurements of fluid loss using various types of plate seals). Agencourt SPRI lids recently became commercially available. Although the SPRI lids are an added cost to the process, they may be reused for up to 10 thermal cycling reactions, and their cost is offset from the savings achieved by reduced volume of sequence reaction mix.
Sealed plates were thermocycled using Tetrad peltier thermal cyclers (MJ Research). Thermal cycling conditions for the reaction format presented here were 50 × (96°C, 10 sec; 43°C, 5 sec; 60°C, 240 sec) with ramping rates of 1°C/sec. The optimal number of cycles was not evaluated, and fewer than 50 cycles may be adequate. Unincorporated nucleotides were removed from the sequence reactions by ethanol/EDTA precipitation as described in Yang et al. (2005), with the exception that 6.6 μL of 38 mM EDTA pH 8, rather than 2 μL of 125 mM EDTA pH 8, was added to the sequencing reaction products prior to the addition of 18 μL of 95% EtOH.
Purified sequence reaction products were resuspended overnight at 4°C (to maximize recovery of purified reaction products) in 10 μL Ultrapure Water (Invitrogen) and sequenced on one of seven 3730xl DNA Analyzers (Applied Biosystems) using 50-cm capillaries and POP-7 polymer (Applied Biosystems). Because the sequence reaction products from our nanoliter-scale reactions were resuspended in the same volume as our standard 4-μL reactions, the labeled DNA was at lower concentration. As such, it was necessary to optimize the electrokinetic injection parameters such that a sufficient amount of labeled reaction products was injected in each capillary electrophoresis run. For the standard Applied Biosystems 3730xl run module, it was empirically determined that doubling the injection time (to 30 sec) but keeping the injection voltage, run time, and run voltage the same (1.5 kV, 5640 sec, and 8.5 kV, respectively) loaded sufficient material into the capillaries of the sequencers for generation of equivalent read lengths to our standard 4-μL reactions. The PHRED software package (v 0.020425.C) (Ewing and Green 1998) was used for base-calling and quality score assignments. For each read, the reported length is a count of the total number of PHRED20 bases.
Acknowledgments
We thank Jim Kronstad, Pieter J. de Jong, Marian Sadar, Robert Brunham, and Vancouver, B.C., Canada and IMAGE Consortium for DNA libraries used in this study. We thank members of the DNA Sequencing and Informatics Groups at Canada's Michael Smith Genome Sciences Centre for their technical assistance. In particular, we thank Ranibar Guin and Joseph Ray Santos for help with data processing. M.A.M. and R.A.H. are Michael Smith Foundation for Health Research scholars.
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4221805. Article published online before print in September 2005.
-
↵3 Corresponding author. E-mail rholt{at}bcgsc.ca; fax (604) 877-6085.
-
- Accepted August 1, 2005.
- Received June 1, 2005.
- Cold Spring Harbor Laboratory Press








