
Methylation-Spanning Linker Libraries Link Gene-Rich Regions and Identify Epigenetic Boundaries in Zea mays
Abstract
Complex cereal genomes are largely composed of small gene-rich regions intermixed with 5 kb to 200 kb blocks of repetitive DNA. The repetitive DNA blocks are usually 5-methylated at 5′-CG-3′ and 5′-CNG-3′ cytosines in most or all adult tissues, while the genes are generally unmethylated at these sites. We have developed methylation-spanning linker library (MSLL) technology as a tool to span large methylated DNA blocks and thereby link unmethylated genic regions. MSLL clones contain insertions of large fragments that are size fractionated over gels after complete digestion of total genomic DNA with restriction enzymes that are sensitive to the 5-methylation of cytosines in 5′-CG-3′ and 5′-CNG-3′ sequences. Our data indicate that the end sequences of maize MSLL clones are greatly depleted in repetitive DNAs and enriched in genes relative to total genomic DNA. Combined with other gene-enrichment approaches, MSLL technology can efficiently generate fully-linked contiguous sequences in complex genomes that are resistant to shotgun sequencing.
Large grass genomes, including those of barley, maize, and wheat, are mostly comprised of 5–20 kb blocks of genes intermixed with repetitive DNA blocks that range in size from a few kb up to more than 100 kb (SanMiguel et al. 1996; Panstruga et al. 1998;Tikhonov et al. 1999; Dubcovsky et al. 2001; Wicker et al. 2001). In a few cases, including within tandem gene families or the rare unrelated gene cluster (Llaca and Messing 1998; Fu et al. 2001), gene-rich regions may extend for 50 kb or more. In most cases, however, gene-rich chromosome segments only contain 1–4 genes in a region of 20 kb or less. The repetitive DNAs found in the intermixed repeat blocks are usually nested insertions of a class of mobile DNAs called long terminal repeat- (LTR-) retrotransposons (SanMiguel et al. 1996; Llaca and Messing 1998; Panstruga et al. 1998; Kumar and Bennetzen 1999;Tikhonov et al. 1999; Dubcovsky et al. 2001; Fu et al. 2001; Wicker et al. 2001). These nested LTR-retrotransposons can make up well over 50% of total genomic DNA, with most of this DNA coming from only a handful of different element families that have copy numbers of several thousand per nuclear genome (SanMiguel and Bennetzen 1998; Kumar and Bennetzen 1999; Vicient et al. 1999; Meyers et al. 2001). The LTR-retrotransposons are relatively large in size (usually greater than 5 kb) and can have numerous copies that are < 99% identical within the same genome (W. Ramakrishna, P. SanMiguel and J. Bennetzen, unpubl. obs.). Hence shotgun sequencing of complex grass genomes would not yield information that can be converted into long contiguous sequences (Bennetzen et al. 2001). Nuclear genomes in higher plants contain extensive 5-methylation of cytosine residues, much of it associated with 5′-CG-3′ and 5′-CNG-3′ sequences (Gruenbaum et al. 1981a). In many animals and plants, cytosine methylation is associated with heterochromatic regions, where it apparently contributes to the transcriptional inactivity of any sequences within the condensed chromatin. In maize, studies indicate that most of this cytosine methylation is associated with repetitive DNAs, including the LTR-retrotransposons. In adult tissues, most LTR-retrotransposons appear to be 100% methylated at all 5′-CG-3′ and 5′-CNG-3′ sites, while genes appear to be unmethylated at these same sites (Gruenbaum et al. 1981b); Antequera and Bird 1988; Bennetzen et al. 1994). This lack of genic methylation differs somewhat from that observed in mammals (Bird 1986; Frank et al. 1991), for instance, because the absence of methylation is even found in genes that are not expressed in the tissues that were the source of the DNA that was characterized (Bennetzen et al. 1994). There are likely to be exceptions to this general rule (Jacobsen and Meyerowitz 1997), but overall it appears that most genic regions can be separated from most LTR-retrotransposon blocks by this difference in DNA methylation. Perhaps most interesting of all, the size of the methylated DNA blocks has an upper limit of 200 kb or less (Springer 1992; Bennetzen et al. 1994), suggesting that an 'open' region of chromatin is needed at this spacing to allow some essential nuclear function, such as the initiation of DNA replication or chromosome folding.
Martienssen and coworkers have used the difference in DNA methylation between repetitive and genic DNA as a tool to efficiently sequence gene-rich regions of the maize genome by a shotgun approach (Rabinowicz et al. 1999). In their methyl filtration technology, the insertion of sheared fragments of total genomic DNA into a plasmid vector is followed by transformation of this library into an Escherichia coli strain that will not tolerate 5-methylation of cytosines in the cloned DNA. In maize, this approach yielded a greater than two-fold enrichment for genic sequences and at least a six-fold depletion of known LTR-retrotransposon sequences, relative to the same library inserted into a methylation-tolerant E. coli host (Rabinowicz et al. 1999; Meyers et al. 2001). Application of methyl filtration technology to the full maize genome should yield contiguous sequences (contigs) for the genic regions, varying in size from a few kb up to a few dozen kb. However, this filtration technique does not localize the genic contigs relative to each other or to the maize genetic map. We have developed an approach that we call methylation-spanning linker library (MSLL) technology that overcomes this deficiency and also isolates the boundaries between methylated and unmethylated regions.
RESULTS AND DISCUSSION
Figure 1 depicts the structure of a contiguous 225 kb of maize nuclear DNA that contains theadh1-F locus, as derived from the sequence of Tikhonov and coworkers (Tikhonov et al. 1999). Subsequent studies have shown that this is the standard structure for most or all gene-containing portions of the genome of maize and other large-genome cereals such as wheat and barley (Edwards et al. 1992; Llaca and Messing 1998; Dubcovsky et al. 2001; Fu et al. 2001; Wicker et al. 2001). These genic regions contain small islands of genes separated from each other by relatively large blocks of repetitive DNA. Because these repetitive DNA blocks are cytosine 5-methylated at all 5′-CG-3′ and 5′-CNG-3′ sites, they will not be digested by restriction enzymes that are inhibited by this type of DNA methylation. The lines in Figure 1 show the fragments that would be generated by complete digestion with SalI orHpaII, two enzymes that are cytosine-methylation sensitive.
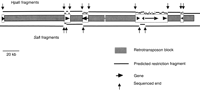
Structure of the maize adh1-F region, an example of a genic segment of the maize genome. Narrow shaded boxes indicate blocks of methylated DNA consisting of nested retrotransposons (SanMiguel et al. 1996). The open boxes indicate gene islands, with genes depicted as arrows or arrowheads oriented in the predicted direction of their transcription and with a length indicative of their predicted initial transcript size. The lines indicate the predicted HpaII (top) or SalI (bottom) fragments that would be generated from digestion of maize genomic DNA if all of theHpaII and SalI sites in the retrotransposon blocks are 100% methylated at all 5′-CG-3′ and 5′-CNG-3′ sites, while these sites are 100% unmethylated in the gene islands. Vertical arrows indicate MSLL ends that would be sequenced. Data taken from Tikhonov (Tikhonov et al. 1999).
Because HpaII has a 4 bp recognition/cleavage sequence, it should cut maize DNA, which has an approximate 50% GC content (Hake and Walbot 1980), an average of once every 44 (256) bp. However, from the analysis of bulk maize sequence data (Meyers et al. 2001), we found that the maize genome is about 53% AT, also observing that 5′-CG-3′ and 5′-CNG-3′ are somewhat underrepresented in the maize genome (a respective 4.6% and 5.3%, compared to the predicted 5.6%). In accord with the depletion of these bases, the actual frequency ofHpaII sites that we detected in the bulk sequence data generated by Meyers et al. (2001) was once in every 305 bp. However, pulsed field gel analysis of HpaII-digested maize DNA indicates that the majority of the genome yields fragments that are larger than 50 kb (Springer 1992; Bennetzen et al. 1994). Hence isolation of HpaII fragments larger than a few kb should yield segments of DNA that contain internal methylated and repetitive sequences, while the ends are anchored in the unmethylated sequences associated with genic regions. Sequencing the ends of such fragments will mark adjacent genic regions, and provide clones that contain the sequences between those two genic regions. SalI can be used in the same manner, although its 6 bp specificity indicates that it will digest (on average) several kb away from the site at which a methylated DNA block begins.
To test this theory, we constructed three small BAC libraries. One library contained 9–14 kb HpaII fragments in the vector pBeloBAC11 (Kim et al. 1996). The fragments were generated by complete digestion of B73 maize genomic DNA with HpaII, followed by pulsed field gel electrophoresis of the digested DNA. Fragments of 9–14 kb were excised from the gel, half-filled (Zabarovsky and Allikmets 1986) with dCTP, and ligated into the vector digested with BamHI and partially filled with dATP, dGTP and dTTP. The second two libraries were similarly constructed except that they used fragments from a complete SalI digestion, half-filled and ligated into BamHI-digested and half-filled pBeloBAC11. The inserts in the SalI libraries were in the size ranges of 10–15 kb and 15–25 kb. The half-fill ligation approach and the choice of fragment size ranges that differed by less than two-fold were both designed to minimize the possibility of chimeric clones.
Both ends of 192 BAC clones from the HpaII library and 96 clones for each of the two SalI libraries were subjected to DNA sequence analysis. The length distribution of the obtained sequences was 100–759 bp, generating 410 kb of total sequence, with a mean read of 545 bp and a median read of 589 bp. Overall, just over 77% of the sequencing reactions yielded 100 bp or more of high quality (PHRED 20) (Ewing and Green 1998) sequence. These sequences were scored for the presence of genes, LTR-retrotransposons, other repeats, and organellar DNA. The same analysis was performed on 167 sequences that we generated for the ends of EcoRI BACs from maize inbred B73 (http://www.chori.org/bacpac). Table1 shows the summarized results for these four sets of data. The HpaII library yielded end sequences that exhibited homology to genes, retrotransposons, and chloroplast DNA for a respective 5%, 25% and 4% of the clones. The end sequences of the smaller SalI library yielded these same classes in a respective 14%, 23%, and 17% of clones, while the largerSalI library yielded a respective 18%, 18%, and 5% for these homologies. In sharp contrast, the EcoRI library yielded ends that were homologous to genes, retrotransposons, and chloroplast DNA for a respective 1%, 52%, and 1% of the time. The results for the EcoRI BAC ends are very similar to those seen in random sequencing of sheared fragments of the maize genome, which gave these same homologies in about 1%, 48%, and 1% of the sequenced clones (Rabinowicz et al. 1999; Meyers et al. 2001).
BAC End Sequence Compositions
These results indicate that the MSLL clones are enriched for genes and deficient for LTR-retrotransposons at their ends. Even though the LTR-retrotransposons are underrepresented in the MSLL libraries, we were surprised that so many LTR-retrotransposon homologies were still detected. However, closer inspection of the sequence data indicated that just over 10% (14/136) of the sequence homologies to LTR-retrotransposons began within the first 10 bp of sequence for the MSLL clones. For the EcoRI library, about 55% (42/77) of the homologies to LTR-retrotransposons were found to begin in the first 10 bp of sequence. Hence many of the MSLL clones have ends that are outside, but very near, LTR-retrotransposon blocks. Because the averageHpaII site will be less than 300 bp from the first methylated region, we expect that this close juxtaposition of the cleaved site and an LTR-retrotransposon block should be particularly frequent inHpaII-based MSLL clones.
Of the 292 HpaII sequences that gave 100 bp or more of PHRED 20 sequence, 106 sequences were found to have one or more additionalHpaII sites within the sequences generated. Many of theseHpaII sites (71%) are within annotated LTR-retrotransposons. Despite their extensive methylation at 5′-CG-3′ and 5′-CNG-3′ sites (Bennetzen et al. 1994), our sequence inspections have shown that the most abundant LTR-retrotransposons that make up over half of the maize genome (SanMiguel and Bennetzen 1998; Meyers et al. 2001) are actually enriched in HpaII sites relative to genes (one per 239 bp versus one per 492 bp for genes), largely because of the higher average GC content of the LTR-retrotransposons.
Various crude predictions for the maize genome suggest that around 5%–15% of the total nuclear DNA is composed of genes, while about 50%–80% is composed of LTR-retrotransposons (SanMiguel and Bennetzen 1998; Meyers et al. 2001). Part of the rationale for using an internal control with EcoRI clone ends was to balance our criteria for gene and LTR-retrotransposon identification. We expect that the true frequencies of both genic and LTR-retrotransposon sequences in our data sets are higher than the conservative numbers that we apply, but we used the same criteria for both MSLL and EcoRI clones. Because the predicted distance between the cleaved HpaII site and the LTR-retrotransposon block should average less than 300 bp, we expect that HpaII BAC end sites will rarely be within the peptide-encoding portion of a gene, and thus rarely identified as genic.
The frequency of chloroplast DNA homologies in our MSLL clones was about as expected, given that these libraries contained relatively small inserts. In the SalI libraries, for instance, the same chloroplast fragments were seen over and over again, and these were the rare chloroplast SalI fragments that were of the appropriate size to be found in these libraries. Libraries made with biggerSalI fragments, larger than 50 kb, should not have any organellar DNA fragments. These chloroplast DNA fragments were useful, however, in that they exhibited the expected chloroplast sequence homology at both ends, suggesting that the libraries did not have many chimeric clones.
The results indicate that MSLL technology can be used to link adjacent genic regions, while providing the intervening repetitive/methylated DNA block on a clone that is available for any subsequent analysis. In order to be comprehensive, several complete digestion libraries would need to be made across a large range of DNA sizes and with a variety of restriction enzymes. HpaII would be especially efficient for spanning small repetitive DNA blocks (those less than 15 kb or so), whereas SalI, PstI, SmaI, SstII or other methylation-sensitive enzymes with a 6 bp specificity could best characterize large methylated blocks. End sequences of these BACs would link and order all unmethylated regions. Combined with the sequences of these unmethylated regions by methyl filtration shotgun sequencing (Rabinowicz et al. 1999), the MSLL data would permit the assembly of full chromosome contigs.
The sequences of the MSLL BAC ends identify the boundaries between unmethylated DNA (e.g., the cleaved HpaII site) and methylated DNA (e.g., the first HpaII site in the BAC end sequence, hence the first methylated HpaII site). It is not known how these “epigenetic boundaries” are composed in plants, how they are established, or what effects they may have on adjacent genes. The MSLL technology provides comprehensive access to these regions, making them available for more detailed study.
Although our experiments were focused on characterization of the maize genome, MSLL technology should be equally useful for application to any genome with a structure similar to that of maize. These similarly accessible genomes would certainly include barley, wheat, and numerous other vascular plants, but could also include many animals, protests, or fungi with complex genomes and DNA methylation that is enriched in repetitive DNAs.
METHODS
Preparation of High-Molecular-Weight (HMW) DNA From Maize
Maize inbred B73 seeds were kindly provided by Dr. Chris Staiger (Purdue University). HMW DNA was extracted from the leaves of 10-day-old seedlings, as previously described (Liu and Whittier 1994). The final nuclear pellet was embedded in an equal volume of 1.5% low-melting-point agarose. Plugs containing 4–5 ug of DNA were treated with lysis buffer (1% sodium lauryl sarcosine, 0.1 mg/ml proteinase K, 0.1% ascorbic acid, 0.5M EDTA pH 9.1) in 50 ml volume for 48 h at 50°C, with one change of lysis buffer after 24 h.
Digestion, Size Selection of HMW DNA, and BAC Library Construction
Before digestion, the agarose plugs were washed at 50°C in several volumes of washing buffer containing 1mM phenylmethylsulfonyl fluoride (PMSF). 6–8 plugs were equilibrated for 30 min to 1 h in 400 ul SalI or HpaII buffer. Digestion was performed in 200 ul volumes with 60 units of restriction enzymes at 37°C for 12–14 h to achieve complete digestion. Digested DNA plugs were size fractionated on the CHEF-DRII system (Bio-Rad) and visualized under DARK READER (Clare Chemical Research). Different size fragments were cut from the gel and recovered by GElase (Epicentre Technologies).SalI fragments were half filled with dTTP and dCTP, and ligated into the pBeloBAC11 vector digested with BamHI and partially filled with dATP and dGTP. HpaII fragments were partially filled with dCTP and cloned into the same vector digested with BamHI and partially filled with dATP, dGTP, and dTTP. Ligations were transformed into ElectroMAX DH10B-competent cells (Life Technologies).
BAC End Sequencing and Analysis
The BAC DNA templates were prepared following a modification of the standard procedure (Kelley et al. 1999). In brief, a 96-well block containing 1.3 ml per well of LB medium with chloramphenicol was incubated at 37οC for 16 h. 100 ul of the overnight culture was transferred to four 96-well blocks containing 1.3 ml per well of LB medium with chloramphenicol, and grown at 37οC for 14 hr. The BAC DNA was isolated from each block using the Qiagen R.E.A.L. prep 96 system following the manufacturer's instructions. The final DNA pellet containing the pooled DNA from each of the four identical cultures was dissolved in 40 ul of water. Sequencing reactions were set up with 10 ul template, 6 ul 5 X ABI (Perkin Elmer), 4ul Big Dye (Perkin Elmer), 1 ul DMSO and 0.1 ul forward or reverse universal primer in a final reaction volume of 21.1 ul. The sequence traces were transferred to a Sun E450 server and bases were called using phred (Ewing and Green 1998). Vector sequences were masked by CROSS_MATCH. BLAST (Altschul et al. 1997) was employed to compare all the trimmed sequences with the public sequence database as both nucleotides and predicted amino acid translations. Retrotransposons were detected additionally by CROSS_MATCH and TBLASTX against a set of 103 known retroelements.
WEB SITE REFERENCES
http://www.chori.org/bacpac; BACPAC Resources home page for Pieter de Jong's lab at the Children's Hospital Oakland Research Institute.
Acknowledgments
We thank the US National Science Foundation for support of this project (grants 9975618 and 9975793).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL maize{at}bilbo.bio.purdue.edu; FAX (765) 496-1496.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.185902.
-
- Received February 14, 2002.
- Accepted July 17, 2002.
- Cold Spring Harbor Laboratory Press








