
What is Finished, and Why Does it Matter
Our ability to acquire and analyze DNA sequence data has increased phenomenally in the past 12 years. The acquisition of both cDNA and genomic DNA sequence has exerted a major influence on the direction of biological and medical research and will continue to do so. However, the DNA sequencing field has progressed so rapidly that technical differences between various sequencing approaches have resulted in large datasets of differing quality. Although all of these datasets are valuable in their own right, they are composed of experimental data; therefore they are subject to errors, ambiguities, and incompleteness at a level related to the experimental strategy that created them. The picture is further complicated by the lack of a community-accepted nomenclature that clearly defines levels of sequence completeness. Because of the small number of people producing this resource relative to the large number using it, the nature of the data is, unfortunately, not commonly appreciated.
Initially, DNA sequencing was targeted at small (less than 5 kb) genomic regions or cDNAs; thus, there were fewer than 10 sequences of >50 kb available in public databases until the late 1980's (GenBank). This early period established in many peoples' minds the definition of a finished sequence; namely, if a sequence contained no gaps or ambiguities (only A, T, G, and C), then the sequence was complete and accurate (usually as measured by a correct translation to a known protein). As large genome projects were getting underway, this definition became inadequate. For example, no one was planning to sequence human centromeres when sequencing the human genome was discussed. Moreover, the nature of data collection made much of the scientifically applicable information in a DNA sequence available before reaching the level of finished high quality sequence (McCombie et al. 1992). Thus a valuable but less than complete or highly accurate dataset could be provided in a much more timely and less costly manner. These factors further blurred any definition of finished sequence.
Large-scale sequencing progresses in two distinct phases. First is the high-throughput, random data collection or shotgun phase that occurs regardless of whether a whole genome or clone-based approach is used. The data are then ‘assembled‘ using one of several algorithms written for either whole-genome or single-clone assembly. The algorithms overlap the individual sequence reads on the basis of sequence similarity. These programs may also incorporate information from the paired-end data of sequenced double-stranded clones to provide a higher degree of structure and order to the assembly. This results in the accumulation of 80% to nearly 100% of the original DNA sequence, typically contained in small, nonordered assemblies or “contigs.” In the case of map-based sequencing, however, the work spent mapping allows the high-throughput, random data to be assembled in bins according to its clone of origin, thus providing a higher level of structure to the data as well as a rigorous confirmation that the final sequence assembly is correct.
The next step in the large-scale sequencing process is often referred to as “finishing.” In this step, contiguous segments of sequence are ordered and linked to one another and any ambiguities or discrepancies among the individual reads are resolved. Once this is concluded, a relatively rigorous quality check and verification is performed. At this stage any suspicious assemblies are analyzed and either verified or disassembled. In some projects (e.g., portions of chromosome 10 of rice), up to 30% of the initial assemblies may be inconsistent with other data such as restriction digest fingerprints (M. Delabastide and W.R. McCombie, pers. comm.), and a finishing stage is critical to the usefulness of the final data. This finishing step adds considerably to the cost and time required to sequence a genome yet provides a level of contiguity and error checking not otherwise possible. The ambiguity in what is finished and what is not stems from the disassociation of these two major sequencing steps in large-scale sequencing projects. In addition, differing degrees of random coverage lead to differing qualities of incomplete sequence. Simply put, all things being equal, sequence with more random coverage will be represented in larger contigs of higher quality than sequence with a lower degree of coverage. Moreover, the issue of what is finished is further complicated by the fact that even a “finished” genome is rarely complete when one is considering higher eukaryotes.
Although several finished bacterial genomes are truly complete (they are represented by a single contiguous sequence with no ambiguities), such is not the case for multicellular organisms. Caenorhabditis elegans approaches such a level of completion as a result of the massive effort put into the project, as well as its lack of centromeres ( C. elegans Sequencing Consortium 1998). Other “complete” genomes such as Arabidopsis thaliana do have regions without sequence data. For example, the centromeres of Arabidopsiscontain a core repeat that is refractive to current sequencing technology (Arabidopsis Genome Initiative 2000). Thus by a strict definition, no genome of a multicellular organism is completely finished. However, the sequence that is available is finished to a high quality and represents the genome in large contiguous segments. Clearly, there are differences among the various complete (yet incomplete) genomes. At the time of its publication, the 125-mbArabidopsis genome was represented by 6 of 10 chromosome arms that were contiguous from telomeric repeat to centromeric repeats (Arabidopsis Genome Initiative 2000; R. Martienssen, pers. comm.). One of the remaining chromosomes had gaps of partially sequenced pericentromeric heterochromatin. The other had gaps in the pericentromeric heterochromatin region as well as a few gaps (fewer than five) in difficult-to-sequence regions in other areas. About half of the estimated heterochromatin was sequenced. This adds up to a genome with 15–20 gaps, which is approximately one gap per 8 million bases. Drosophila melanogaster, in contrast, has about 1600 gaps in a genome of similar size, yielding about one gap per 75,000 base pairs (Adams et al. 2000). Figure 1 is a histogram that displays the number of gaps per kilobase for several recently completed genome projects. The numbers used were described in the initial publications, and all of the genomes have likely progressed toward completion in the intervening time; for example, only one gap of the initial 146 reported remains in the C. elegans sequence (R. Wilson, pers. comm.).
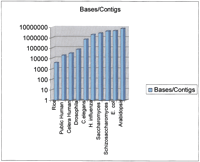
The figure shows the results of dividing the genome size by the number of contigs in the sequence at the time of initial publication. Although other ways to measure contig size are often used, such as N50, they were impossible to estimate on all of these projects. As a result we used this simpler metric, which provides a reasonable estimate of the degree of continuity in the sequence. Genomes represented and the source of the data are: Rice (indica) publicly available draft sequence (Yu et al. 2002); public human genome (Lander et al. 2001); Celera human genome (Venter et al. 2001); Drosophila melanogaster(Adams et al. 2000); Caenorhabditis elegans (The C. elegans Sequencing Consortium 1998); Haemophilus influenzae (Fleischmann et al. 1995); Saccharomyces cerevisiae (Saccharomyces Genome Database,http://genome-www.stanford.edu/Saccharomyces); Schizosaccharomyces pombe (Wood et al.); Escherichia coli (Blattner et al. 1997); and Arabidopsis thaliana (Arabidopsis Genome Initiative 2000).
What is clear from Figure 1 is the variable degree of completion of these published genomes, expressed as the number of bases per contig, although clusters are evident. Some organisms (Arabidopsis, C. elegans and bacteria) are in a group that is largely or exclusively represented by contigs that range from hundreds of thousands up to millions of bases. The second group has contigs in the thousands to tens of thousands of bases (rice, human,Drosophila). Some of this is attributable to the biology of the organism, but the sequencing approach taken markedly impacts the result. In some cases (e.g., the publicly produced human and rice sequences) the sequence at the time of publication is clearly stated as being of draft quality, that is, a work in progress (Lander et al. 2001, Goff et al. 2002, Yu et al. 2002). In these cases, the speed and cost savings possible led the sequencing groups to dissociate the production and finishing steps and to publish an assembly of the random shotgun sequence data that is quite valuable to the respective research communities.
One unfortunate side issue of this practice has been that certain publications describing sequencing projects of this type have not clearly defined their valuable contributions as draft-quality sequence (Venter et al. 2001; Adams et al. 2000). This has the unfortunate effect of leading end-users to underestimate the limitations of the datasets. It can also interfere with the impetus to carry forward and finish the sequence—but, in fact, why should the sequence be finished?
Draft sequence has incredible value for a variety of studies because most genes are represented in the draft sequence of an organism. Even if virtually all genes are present on multiple fragments in the draft, a competent experimentalist can piece together and verify sequences of interest in short order. Draft sequence can also provide a comprehensive estimate of the number of genes, their classifications, and their relatedness to the gene sets of other organisms.
The limitation of draft sequence is a result of two main shortcomings. One of these, the relative lack of contig order, is simpler to understand. Although draft sequence often has order information attached to it, this is less comprehensive than that associated with complete sequences. The order information fails on several levels in draft sequences. On the largest scale, the orientation of the scaffolds (or supercontigs) to one another is often not clear. On the smaller scale, the orientation and sometimes the order of the contigs within a scaffold is ambiguous or in error.
Two additional problems result from this lack of order. Although it may be relatively efficient to order fragments and fill gaps in a single gene, this requires time and money. One of the reasons the first genome projects were initiated was the understanding that completing genes individually would be relatively inefficient, whereas completing all of the genes together would provide substantial efficiency and cost savings. Having the community complete 25,000 genes inArabidopsis, for instance, from a draft sequence would have cost far more and taken significantly longer than having it performed by the consortium that sequenced the organism.
The other point related to the lack of order in the draft is the difficulty in using draft sequence as a reference in global genome and comparative studies. This is of particular importance in the case of the rice genome. Rice is at the center of a field of syntenic cereal (corn, wheat, barley, oats) genomes (Gale and Devos 1998). These genomes represent many important crops, and all of them have significantly larger genomes; hence rice represents a master reference point for the genomes of cereal crops. The lack of completion in the rice genome has serious implications for its use as a reference in comparative genome analysis. Once the reference genome has been finished, this burden is substantially lifted from the remaining related genomes that are to be sequenced. For example, the rice sequence can be used to order contigs of maize sequence from the same mapping bin. However, deletions and insertions of additional genes in rice or maize mean that this bin will need to be more completely finished in maize before the sequence is of use. Typically this would be performed by PCR amplification of gaps between contigs from underlying BAC clones, but other methods are under development. Clearly, such an approach would be impossible without reference to a standard, finished genome such as rice. Comparing two partially completed genomes, although valuable for gene discovery, would soon lead to potentially compromised genetic experiments in the absence of a good map.
The last limitation of a draft versus “complete” sequence relates to the growing distinction between genomics and genetics. One of the major intellectual distinctions of genome analysis versus other biological studies is its potential for completion. A complete genome sequence makes the fundamental hereditary content of an organism finite. There are of course error bars on this as with any experimental data, but those error bars are far greater in a draft genome than in a finished sequence. Examining all rather than most of the genes in an organism (how they are physically localized in the genome and how the structural characteristics of the genome regulate function and inheritance) requires a finished sequence. Knowing which genes are not found in a particular branch of the evolutionary tree has enormous implications and is equally important. Although this is of obvious importance in microbial genomes, the growing realization that this is the case in higher organisms is beginning to have profound effects on areas such as cancer and its models. For genetic experiments involving a particular gene, a draft sequence will likely suffice. Studying the function and transmission of the complete hereditary information of an organism requires the finished sequence of that organism or a closely related reference sequence. In summary, finished data of the highest quality is the most desirable state for a genome sequence, but draft quality sequence can provide a powerful resource for many genomic experiments. What is important is that the end-user has a realistic understanding of the data quality and the implications associated with that quality.
WEB SITE REFERENCES
http://genome-www.stanford.edu/Saccharomyces; Saccharomyces genome database.
Footnotes
-
↵3 Corresponding author
-
E-MAIL McCombie{at}cshl.edu.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.032102.
- Cold Spring Harbor Laboratory Press








