
Why Names
And Adam gave names to all cattle, and to the fowl of the air, and to every beast of the field.Genesis 2:20
Complexity and the Self-Similar Nature of Evolutionary Trees
Description may easily be a never-ending task if the amount of desirable detail is not specified beforehand. The endless complexity of nature is ingrained in the fractal geometry of organisms (such as corals) or their parts (such as lungs), but the fractal properties of the basic structure of life are also apparent in organelles, biochemistry, and genomes. That is why one of the most ubiquitous fractal shapes in nature, the tree, is used in many biological fields, and quite prominently in evolution. It allows the portrayal of the general shape of a branching process without reference to its most minute details. If a tree is constructed from the information in a genome segment, adding further information will sprout a new, self-similar tree, at the preexisting branches. This could proceed ad infinitum until all individual genomes have been sequenced in their entirety, and it would continue to grow in future generations.
Clearly, it is not the case that the more complexity, the better, and it seems that we should devise ways of extracting meaningful insights from what may become a dizzyingly complex tree. Here we suggest such a two-tiered strategy:
1. Fix a departing point in the past time where all extant variation coalesces for a species and a given genome region. Obviously, this past trunk was itself in the midst of a thicket, but all those other branches have become extinct and are irretrievable unless we study all past organisms.
2. From that departing point in the past, follow forward in time until the tree has grown to the desirable (manageable) bushiness or complexity. This level falls far short of the individual information that we have at the actual tips of the tree. We need to prune the tree without creating topiary figures, that is, to reduce the complexity without altering its overall shape, because the shape of the tree contains information regarding both its completeness and the evolutionary processes that created it. We will consider these two aspects of the shape of a tree in relation to the human Y chromosome tree presented in this issue by the Y Chromosome Consortium (YCC) (2002).
A Tree of Trees
The discovery of polymorphisms in the Y chromosome, particularly of PCR-typeable biallelic markers, lagged clearly behind the rest of the chromosomes. However, the field exploded a few years ago, with different laboratories developing their own sets of unique evolutionary polymorphisms (UEPs, an umbrella term that encloses SNPs and indel polymorphisms, and stresses their slow mutation rate compared to minisatellites and microsatellites; de Knijff 2000). Thus, many reports have begun by listing the UEPs typed, the names of the haplotypes resulting from the combination of those UEPs, and the tree that linked them. Recombination is absent in most of the Y chromosome, that is, in the self-describing non-recombining region (NRY). Thus, the whole NRY has had a single evolutionary history, and the different trees presented should be sketches of the overall underlying tree. Direct comparison of the different UEP sets in order to reconstruct this single tree was difficult at best, often impossible. Just identifying the correspondences among haplotypes in different sets proved a daunting task. Presented with this situation, the YCC took the best route to solve it: they promoted joint, collaborative research in which samples were distributed among the different laboratories to be typed for each different UEP set. From the joint results, a tree was reconstructed, showing which mutations fell in the same branches and which lineages were, in fact, defined by a unique UEP allele.
The next step consisted of devising a nomenclature system that would describe the haplotype diversity found. This naming system, modeled on the letter codes used in mtDNA, has three interesting properties: (1) it defines phylogeographically meaningful sets of haplotypes (known as haplogroups), with a clearly structured geographical pattern (Fig.1); (2) it specifies in very intuitive ways how the nomenclature system would adapt to future UEP additions to the tree; and (3) it accommodates partial typings, which is not a trivial matter, since the tree contains 237 polymorphic sites and many laboratories will type a selection of those sites; this feature also makes the nomenclature retroactive.
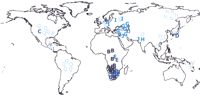
Haplogroups and geographic origins of all individuals in the Y Chromosome Consortium (2002) collection. It comprises the 74 individuals from Table 1 in The Y Chromosome Consortium (2002). Haplogroups are depicted according to the letter proposed as nomenclature by the YCC, with four intensities of color according to their position in the phylogenetic tree (in four decreasing intensity classes: haplogroups A and B, C through E, F through J, and K through R).
Shape and Completeness of the Tree
First, the tree is robust. In a nonrecombining region, phylogeny reconstruction is a straightforward process, up to the point that recurrent mutation at eight out of 237 sites can be recognized unambiguously. With the confidence given by the tree robustness, we shall now proceed to analyze the shape of the tree and to derive conclusions on two fronts, as stated above: the completeness of the tree and the evolutionary forces that shaped it.
Evolutionary trees come in two basic shapes: regular and star-like, which are the result of different evolutionary processes. The shape can be described through the pairwise difference distribution; that is, the number of allele differences between all possible haplotype pairs. A regular tree, with long internal branches, will result in different groups of haplotypes and a multimodal pairwise difference distribution (von Haeseler et al. 1996); in contrast, star-like trees have short internal branches and most of the terminal branches are independent and of similar length. Then, the pairwise difference distribution would appear bell-shaped. The YCC tree is star-like, since its associated pairwise difference distribution is clearly bell-shaped (Fig.2a). Armed with this knowledge, we can now test some hypotheses.
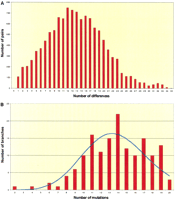
(A) Pairwise difference distribution among all pairs of YCC haplotypes. (B) Branch length distribution, in number of mutations, from the tree root to each tip. Blue line: Poisson distribution. Both parts of this figure are based on the tree proposed by the Y Chromosome Consortium (2002).
In the description of human variation, we may be crippled by a nagging doubt: have we found all of the relevant branches in the tree? How complete is the description? We can adapt these questions to the YCC tree. The number of mutations from the root of the tree to each haplotype should be roughly similar, but, since mutation accumulation is a stochastic process, that number of mutations follows a Poisson distribution. Inordinately short branches can be detected in this way, pointing to missing UEPs. The results (Fig. 2b) show that, at most, two out of 153 branches (those ending in haplotypes A1 and A3a and containing, respectively, two and four mutations) may be unexpectedly short. Thus, we may be reassured that the YCC tree is, in its basic structure, complete.
The YCC Tree and Genomic Context
Next, we can examine a genomic aspect of the process of variation accumulation. The YCC has found eight recurring mutations in the 245 (mostly) UEPs they examined. Is that a chance event? Or, alternatively, do different sites mutate at different rates on the NRY? If a given number of mutation events rains at random on a given genomic length, the number of mutations per site follows a Poisson distribution. Since the whole UEP set was not ascertained from a well delimited genome segment, we must estimate first the length of the Y chromosome that would contain 245 polymorphic sites. Shen et al. (2000) found a density of one polymorphism every 986 bp in 64 Kb of sequence comprising three Y chromosome genes. Then, 245 polymorphic sites would correspond to a length of 241,570 bp, and to an average 1.047 × 10−3mutations per site. Under a Poisson distribution, 244.7 sites would be expected to have mutated once and just 0.124 twice. Since eight sites were found to have mutated twice, this means that not all sites mutate at the same rate. It is not an unexpected result in a coding region, and with contexts such as CpG dinucleotides and some local rearrangements (such as 12f2) prone to recurrent mutation. Moreover, the typing of a preestablished set of markers in a population sample will make it likelier to detect recurrency than if variation in the chromosome were ascertained independently of previous knowledge about the polymorphic status of a given position. In any case, and as stated above, recurrency is no obstacle to obtain a clear-cut reconstruction of the haplotype phylogeny.
The Y Chromosome Phylogeny and Population Forces
A star-like phylogeny results from a sudden expansion in the number of copies of a genome region. This may happen if all the genome expands, that is, in a demographic population expansion, or if just that region expands, propeled by positive selection on a particular variant. In the NRY, absence of recombination means that positive selection on any given site will both expand that particular NRY background and deplete genetic variation on the whole of the NRY.
Signs of population expansion are found throughout the human genome, and the star-like phylogeny of the Y chromosome may be such a sign. This does not preclude the action of selection, and, in fact, some evidence points to a role of selection in shaping variation at the NRY.Shen et al. (2000) found a reduced nucleotide diversity in three genes in the Y chromosome. It must be taken into account that the effective population size of the Y chromosome is one-quarter of that of any autosome, which reduces by four its potential for harboring diversity. Even after correcting for the different effective population size, nucleotide diversity in the NRY remains lower (Bertranpetit 2000). However, the actual magnitude of the footprint of selection seems to show a small role for selection in shaping Y chromosome genetic variation of extant humans (Pérez-Lezaun et al. 1997). This lead should be followed with comparisons with the homologous ape sequences that may point to relevant functional differences that may have been selected for.
Future Directions
The YCC tree is a starting point rather than an end; it is a powerful tool for exploring population histories at several geographical levels. At the same time, it may grow as new UEPs are incorporated, which is likely to happen if a wider array of populations are screened for new UEPs or if additional NRY segments are sequenced in several individuals. The YCC has devised a naming system both coherent and flexible, which we urge all workers to adopt and all journal editors to enforce.
We would like to end by pointing out that the YCC achievement springs from its collaborative nature and from the participants' determination to pool efforts. This should be a main trend in future genome research.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL jaume.bertranpetit{at}cexs.upf.es; FAX 34-93-542-28-02.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.226502.
- Cold Spring Harbor Laboratory Press








