
Microsatellites in the Eukaryotic DNA Mismatch Repair Genes as Modulators of Evolutionary Mutation Rate
All “minor” components of the human DNA mismatch repair (MMR) system—MSH3, MSH6, PMS2, and the recently discovered MLH3—contain mononucleotide microsatellites in their coding sequences. This intriguing finding contrasts with the situation found in the “major” components of the DNA MMR system—MSH2 and MLH1—and, in fact, most human genes. Although eukaryotic genomes are rich in microsatellites, nontriplet microsatellites are rare in coding regions. The recurring presence of exonal mononucleotide repeat sequences within a single family of human genes would therefore be considered exceptional.
A study of 10,000 random primate (primarily human) coding sequences (8.6 million base pairs) showed that the average-sized sequence coding for a human gene is expected to contain approximately 0.1, 0.03, and 0.006 mononucleotide runs of length 7 bp or more, 8 bp or more, and 9 bp or more, respectively (Metzgar et al. 2000). Together, the four minor MMR genes contain three 7-bp runs, four 8-bp runs, and one 9-bp run. By nature of their form and position within the genes, these runs do not appear to be evolutionarily homologous. The probability of finding this many mononucleotide microsatellites by chance in a group of four genes is 3.2 × 10−7(0.43 × 0.124 × 0.024). Controlling for gene length yields a probability of 1.3 × 10−6. Therefore, the human minor MMR genes contain a significant excess of mononucleotide repeats. The probability would be even lower if the calculation included the MED1 (MBD4) gene, considered by some to be another minor MMR gene, which has an exonal 10-bp polyadenosine sequence.
We surveyed MMR gene sequences in organisms that ranged in complexity from prokaryotes to humans and found that the constellation of microsatellites in the coding regions of the minor MMR genes is a general feature among eukaryotes (Table 1). Although in some species, 7-bp mononucleotide runs are sporadically found in the major MMR genes, the longer runs, which are exponentially more mutable, are exclusively present in the minor MMR genes. Microsatellite sequences are exceptionally vulnerable to spontaneous insertion or deletion mutations, and nontriplet microsatellites, when located in coding sequences, are expected to introduce frameshift loss-of-function mutations at high frequency (Moxon et al. 1994). It is a paradox that the MMR system, which limits mutations in microsatellite sequences, would be particularly vulnerable to mutation by virtue of having microsatellites in its own coding regions. In the minor MMR genes, these otherwise rare and generally negatively selected sequences may have evolved by providing a survival advantage for eukaryotic species. What evolutionary advantage could microsatellites provide in the coding regions of the minor MMR genes? We hypothesize that the exceptional density of microsatellites in the minor MMR genes represents a genetic switch that allows the adaptive mutation rate to be modulated over evolutionary time.
Microsatellites in the Coding Regions of the DNA MMR Genes
The demonstration of a surprisingly high frequency of mutator strains among bacterial pathogens appears to illustrate this paradigm, which is an example of the possible advantage of a high mutation rate within a population of organisms subjected to marked changes in environmental conditions (LeClerc et al. 1996). Under adverse experimental conditions, MMR-deficient bacterial mutators outcompete nonmutators and are common in natural and laboratory populations subjected to intensive selection (Rosenberg et al. 1998). Theoretical work also supports the advantage of mutator phenotypes under conditions that require rapid evolution (Sniegowski et al. 1997; Taddei et al. 1997). Does this paradigm apply to eukaryotes? We propose that the existence of microsatellites in MMR genes might be evidence that extends this concept to higher organisms (Fig. 1).
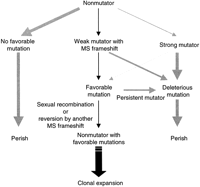
Model of the survival advantage under conditions requiring rapid evolution conferred by the weak mutator phenotype with a microsatellite (MS) sequence in coding regions of the minor DNA MMR genes.
MMR proteins correct replication errors and actively inhibit recombination between diverged sequences (Chen and Jinks-Robertson 1998; Kolodner and Marsischky 1999), thus controlling rates of mutation and evolutionary adaptation. From yeast to humans, the MMR system is composed of homologs of the MutS and MutL genes ofEscherichia coli. Either MSH3 or MSH6 heterodimerizes with MSH2, which creates MutS-α or MutS-β complexes, respectively. Similarly, PMS2 (PMS1 in yeast) heterodimerizes with MLH1 to make MutL-α, and MLH3 is also presumed to combine with MLH1. When the function of either MSH2 or MLH1, the major MMR proteins, is impaired, all MMR activity is lost. However, when one of the minor proteins is impaired, MMR function is retained or only partially perturbed because MSH3 and MSH6 have some functional redundancy and can compensate, at least in part, for the loss of the missing proteins. The case may be similar with PMS2 (PMS1 in yeast) and MLH3. As a result, the loss of activity in any one of these minor proteins generates a weaker mutator phenotype than occurs with loss of the major MMR proteins. A high rate of frameshift mutations that inactivate the minor MMR genes would provide a eukaryotic lineage with a subpopulation of individuals that exhibit mildly increased mutation rates. These individuals would be expected to suffer decreased fitness owing to increased deleterious mutations. Nonetheless, under circumstances favoring rapid evolution, a hypermutable subpopulation would also experience an increase in the rate of necessary adaptive mutations. If these microsatellite sequences had appeared in the coding regions of the major DNA MMR genes, a strong mutator phenotype would be likely to occur; this phenotype might be less capable of overcoming the hazardous effects of the accumulation of deleterious mutations.
When strains acquire “favorable” mutations during a transient hypermutable state, they need to revert to a nonmutator phenotype to keep the newly-obtained adaptive characteristics. Among the strains that have acquired favorable mutations, only those that revert to the nonmutator phenotype are likely to survive. Asexual eukaryotes could achieve the nonmutator phenotype through direct reversion of the loss-of-function mutation, because microsatellite mutations can revert at a high rate. In contrast to other types of mutations, such as transitions or transversions, deletion/insertion mutations within repeated sequences have a higher probability of wild-type reversion, because only one additional cycle of DNA polymerase slippage is required to regain the original sequence. In sexually reproducing eukaryotes, recombination would rapidly separate these adaptive mutations from the (otherwise deleterious) mutator phenotypes with which they would initially be associated. Many of these mutator alleles may in fact be removed by selection, but recombination allows this to happen while simultaneously retaining (or even fixing) beneficial mutations that arose as a result of the mutator alleles' activity. In the minor DNA mismatch repair genes, the microsatellites may therefore act as an evolutionary switch that modulates the mutation rate under conditions that require rapid evolution.
Is the pool of mutator individuals large enough to be relevant in a putative crisis? In evolutionary terms, even a rare, more fit phenotype might be sufficient if these individuals survive and their progeny increase in number. For example, in humans, there is a non-negligible number of MSH6 minor MMR gene germ-line mutations in families with a weakly penetrant form of hereditary colon cancer. Kolodner et al. (1999) estimated that germ-line mutations in MSH6 might be present in 1.5% of all the United States colorectal cancer cases, because familial colon cancer clusters that do not fulfill the Amsterdam criteria for hereditary nonpolyposis colorectal cancer (HNPCC) account for approximately 18% of colorectal cancers, and 8.1% of these probands harbor MSH6 mutations. Moreover, in 4.5% of the familial non-HNPCC probands, the germ-line mutations have been reported to be located in the exonal polycytosine repeat of theMSH6 gene (Shin et al. 1999). Perhaps these families represent a case in point of the process we have described.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL crboland{at}ucsd.edu; FAX (858) 822-0301.
-
Article and publication are athttp://www.genome.org/cgi/doi/10.1101/gr.186301.
- Cold Spring Harbor Laboratory Press








