
Genomic Anatomy of a Premier Major Histocompatibility Complex Paralogous Region on Chromosome 1q21–q22
- Takashi Shiina1,
- Asako Ando1,
- Yumiko Suto2,
- Fumio Kasai2,
- Atsuko Shigenari1,
- Nobusada Takishima1,
- Eri Kikkawa1,
- Kyoko Iwata1,
- Yuko Kuwano1,
- Yuka Kitamura1,
- Yumiko Matsuzawa1,
- Kazumi Sano1,
- Masahiro Nogami1,
- Hisako Kawata1,
- Suyun Li1,
- Yasuhito Fukuzumi3,
- Masaaki Yamazaki3,
- Hiroyuki Tashiro3,
- Gen Tamiya1,
- Atsushi Kohda4,
- Katsuzumi Okumura4,
- Toshimichi Ikemura5,
- Eiichi Soeda6,
- Nobuhisa Mizuki7,
- Minoru Kimura1,
- Seiamak Bahram8, and
- Hidetoshi Inoko1,9
- 1Department of Genetic Information, Division of Molecular Life Science, Tokai University School of Medicine, Bohseidai, Isehara, Kanagawa 259-1193, Japan; 2Department of Biological Science, Graduate School of Science, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan; 3 Bioscience Research Laboratory, Fujiya Co., Ltd., Soya, Hadano, Kanagawa 257-0031, Japan; 4Faculty of Bioresources, Mie University, Tsu, Mie 514-0008, Japan; 5Department of Evolutionary Genetics, National Institute of Genetics, Mishima, Shizuoka 411-0801, Japan; 6Tsu Kuba, Life Science Center, The Institute of Physical and Chemical Research (RIKEN), Yatabe-choh, Tsukuba, Ibaraki 305-0861, Japan; 7Department of Ophthalmology, Yokohama City University School of Medicine, Kanazawa-ku, Yokohama, Kanagawa 236-0004, Japan; 8INSERM-CReS, Centre de Recherche d'Immunologie et d'Hématologie, 67085 Strasbourg, France
Abstract
Human chromosomes 1q21–q25, 6p21.3–22.2, 9q33–q34, and 19p13.1–p13.4 carry clusters of paralogous loci, to date best defined by the flagship 6p MHC region. They have presumably been created by two rounds of large-scale genomic duplications around the time of vertebrate emergence. Phylogenetically, the 1q21–25 region seems most closely related to the 6p21.3 MHC region, as it is only the MHC paralogous region that includes bona fide MHC class I genes, the CD1 and MR1 loci. Here, to clarify the genomic structure of this model MHC paralogous region as well as to gain insight into the evolutionary dynamics of the entire quadriplication process, a detailed analysis of a critical 1.7 megabase (Mb) region was performed. To this end, a composite, deep, YAC, BAC, and PAC contig encompassing all five CD1 genes and linking the centromeric +P5 locus to the telomeric KRTC7 locus was constructed. Within this contig a 1.1-Mb BAC and PAC core segment joining CD1D toFCER1A was fully sequenced and thoroughly analyzed. This led to the mapping of a total of 41 genes (12 expressed genes, 12 possibly expressed genes, and 17 pseudogenes), among which 31 were novel. The latter include 20 olfactory receptor (OR) genes, 9 of which are potentially expressed. Importantly, CD1, SPTA1, OR, and FCERIA belong to multigene families, which have paralogues in the other three regions. Furthermore, it is noteworthy that 12 of the 13 expressed genes in the 1q21–q22 region around the CD1 loci are immunologically relevant. In addition to CD1A-E, these include SPTA1, MNDA, IFI-16, AIM2, BL1A, FY and FCERIA. This functional convergence of structurally unrelated genes is reminiscent of the 6p MHC region, and perhaps represents the emergence of yet another antigen presentation gene cluster, in this case dedicated to lipid/glycolipid antigens rather than antigen-derived peptides.
[The nucleotide sequence data reported in this paper have been submitted to the DDBJ, EMBL, and GenBank databases under accession nos.AB045357–AB045365.]
The 3.6-Mb human Major Histocompatibility Complex (MHC; also known as the Human leukocyte antigen, HLA) on chromosome 6p21.3 is a critical repository for immune response genes. This 230-gene–rich segment has taught us a great deal about immunity as well as about the evolutionary dynamics of compact genomic segments (Campbell and Trowsdale 1997; The MHC Sequencing Consortium 1999;Shiina et al. 1999). Extensive analysis of the genomic organization of the HLA region has revealed that at least 27 of its resident genes possess duplicated copies in at least one of three other restricted regions on chromosomes 1q21–q25, 9q33–q34, and 19p13.1–p13.4 (Sugaya et al. 1994, 1997; Kasahara et al. 1996; Katsanis et al. 1996; Endo et al. 1997; Hughes 1998; Kasahara 1999). ABC transporter gene family members are located on 6p21.3 (TAP1, TAP2), 1q25 (EST31252), and 9q34 (ABC2), proteasome β-type subunit loci can be found on 6p21.3 (LMP2, LMP7) as well as 9q34 (PSMB7), pre-B cell leukemia transcription factors are readily identified on 6p21.3 (PBX2), 1q23 (PBX1), and 9q33–q34 (PBX3), and NOTCHgenes are located on 6p21.3 (NOTCH4), 9q34.3 (NOTCH1) and 19p13.2–p13.1 (NOTCH3). These observations suggest that these four paralogous regions were generated from a common ancestor after two rounds of chromosomal duplication. Moreover, these large-scale duplications possibly enabled at least one of these quadruplicate regions to be relaxed from functional constraints, allowing the formation of the present-day vertebrate MHC, the sophisticated machinery at the heart of the acquired immune system (Abi-Rached et al. 1999). A number of indirect evidences, especially the sequence comparison as well as phylogenetic tree analysis of a number of paralogous genes, allows tracing back these duplicatives events to a common ancestor of jawed vertebrates, from the lineage leading to hagfish and lamprey (Kasahara 1999).
Among the above-mentioned paralogous regions, that of 1q21–q25 is unique because it is the only one outside the MHC carrying divergent, yet genuine histocompatibility-like loci, CD1 and MR1(Albertson et al. 1988; Hashimoto et al. 1995; Riegert et al. 1998). CD1 molecules are cell surface glycoproteins structurally and functionally similar to MHC class I molecules (Calabi and Millstein 1986; Martin et al. 1986). The main difference between these two classes of antigen-presenting loci is indeed their “cargo” peptides in the case of 6p-located MHC class I molecules, and a diverse admixture of glycolipids (issued mainly by various pathogens) in the case of CD1 molecules. This diversification of the presentation capacity of MHC molecules greatly enhances the surveillance capacity of patrolling cytotoxic T cells (Sieling et al. 1995; Burdin et al. 1998). There are five CD1 genes, CD1A to CD1E, originally identified within a 190-kb cosmid segment (Calabi and Milstein 1986; Martin et al. 1987; Calabi et al. 1989; Yu and Milstein 1989). Based on sequence divergence, the CD1 genes can be ordered into three groups: (1) CD1A, CD1B, andCD1C, (2) CD1D, and (3) CD1E (Hughes 1991). Only homologs of human CD1D have been identified in the mouse (Balk et al. 1991), and the rat (Ichimiya et al. 1994). CD1Dmight be a vestige of the ancestral CD1, which plausibly created the present-day human CD1 cluster through sequential duplications (Yu and Mulatein 1989).
Furthermore, paralleling the chromosome 6 HLA region, the CD1 region is of great biomedical importance, as a number of disease-susceptibility loci have been mapped to 1q21–q23; these include genes for elliptocytosis-2, spherocytosis, pyropoikilocytosis (Gallagher et al. 1992), autosomal dominant nonsyndoromic deafness, autosomal dominant nonsyndromic sensorineural 7 (Fagerheim et al. 1996), familial hemiplegic migraine (Ducrons et al. 1997), familial partial lipodystrophy (Jackson et al. 1998), and familial schizophrenia (Brzustowicz et al. 2000). This region has also been implicated in a number of chromosomal translocations; for example t (1; 19) (q23; p13) in lymphoblastic leukemias and t (X; 1) (p11; q21) in papillary renal cell carcinoma (Williams et al. 1984; Weterman et al. 1996).
To clarify the genomic organization of this paralogous CD1 region, and to understand the evolutionary process through which the MHC system acquired its present-day structure, we aimed to establish a comprehensive gene map of a critical 1.7-Mb region. A composite YAC, BAC, and PAC contig was thus constructed, and a core segment of 1.1 Mb encompassing the CD1 genes was completely sequenced. This 1.7-Mb region was found to contain 10 known genes and 31 newly mapped genes or gene candidates including 20 ORctory receptors.
RESULTS
High-Resolution Mapping of the 1.7-Mb Region between the +P5 Site and FCERIA Gene
To clarify the molecular structure and gene organization of a segment of chromosome 1q21–q22 region, paralogous to the MHC and containing the MHC class I-like CD1 genes, we initially PCR-screened YAC, BAC, and PAC libraries with STS and locus/gene-specific primers. As a result, 33 YACs, 8 BACs, and 51 PACs were isolated and assembled into a contig. Their identity was confirmed by Southern hybridizations with clone-derived PCR and EcoRI fragments (data not shown). Below is a description of the cloning and the characterization strategy.
Three YAC clones (800C2, 887G1, and 933H7) containing all of theCD1 genes were isolated from the CEPH YAC library usingCD1C primer pairs (Fig. 1) (Walsh et al. 1996). Each CD1 gene within the clones was identified by PCR using CD1A-E locus-specific primer pairs (Table1). The CD1D and CD1Agenes were included in an additional YAC clone, 367B12. The PAC clone, 810N20, which had a 140-kb insert, contained three CD1 genes:CD1D, CD1A, and CD1C. The CD1B andCD1E genes were included in a telomeric overlapping PAC clone, 893N23. As shown in Figure 1, the order of the five CD1 genes was thus established as CD1D—CD1A—CD1C—CD1B—CD1E from centromere to telomere, spanning ∼190 kb, in accordance with previous predictions (Yu and Milstein 1989). A PCR primer pair for +P5 (D1S3309E), a target binding site for the Wilms' tumor suppressor 1 gene (WT1), mapped previously by two-color fluorescent in situ hybridization (FISH) to 1q21–q22 (Negus et al. 1996), allowed successful amplification from the three previously mentioned YAC clones as well as a fourth one, 367B12. Three additional PAC clones (581I21, 510C21, and 681H10) were obtained by gene-walking using new primer sets corresponding to the telomeric sequence of the PAC clone 747M6 harboring the CD1D and CD1A genes (Fig. 1). The telomeric sequence of the PAC clone 510C21 contained the 5′ end region of the +P5 sequence, which therefore places this locus 200 kb centromeric to the CD1D gene (Fig. 1). Finally, four STS markers (D1S1600, D1S176, WI-8369, and SG4979) were in close proximity to each other, 100 kb centromeric to +P5.
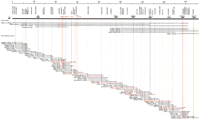
1.7 Mb of a YAC, BAC, and PAC contig between the +P5 and KRTC7 loci. A 1.7-Mb contig constructed by 9 YAC, 8 BAC, and 51 PAC clones is shown with addresses, sizes, markers, and gene contents. BAC and PAC clones subjected to sequencing in Figure 2 are indicated by red letters and lines.
Locus/Gene-Specific Primer Pairs: PCR Primers Used for Screening of Genomic Libraries
Moving stepwise to the telomeric end of this segment, one BAC clone (407I14) and four PAC clones (687N3, 855N14, 987D5, and 1050C4) were isolated using PCR primers designed from the second exon of the erythrocyte alpha-spectrin (SPTA1) gene (Kotula et al. 1991). In addition, the nucleotide sequence at the telomeric end of a PAC clone 1050C4 revealed complete identity with 125-bp overlap to exon 36 of SPTA1. These results showed that SPTA1 was located ∼280 kb telomeric to the CD1E gene (in a telomere-to-centromere orientation) (Fig. 1). Furthermore, the myeloid cell-specific gene, MNDA, was mapped ∼150 kb telomeric toSPTA1 by PCR and Southern hybridization analyses of one BAC clone (407I14) using the MNDA-specific primer pairs and PCR products from the MNDA locus as a probe, respectively (data not shown). Likewise, immunologically relevant genes, the interferon γ-induced gene (IFI-16) (Trapani et al. 1992), and the α subunit of the IgE high-affinity Fc receptor gene (FCERIA) (Kochan et al. 1988) were localized ∼150 kb and 450 kb telomeric to the MNDA gene, respectively. The Duffy blood group antigen locus (FY) was mapped between IFI16 andFCERIA, and the keratinocyte cDNA 7 (KRTC7) gene (Konishi et al. 1994) was charted ∼100 kb telomeric toFCER1A (Fig. 1). All together, we have constructed a high-resolution 1.7-Mb YAC, BAC, and PAC contig between the +P5 and KRTC7 loci. This contig contains at least 14 genes and +P5, in this (centro-telomeric) order: +P5—CD1D—CD1A—CD1C—CD1B—CD1E—SPTA1—MNDA—IFI-16—FY—FCERIA—KRTC7from centromere to telomere (Fig. 1). Finally, representative BACs and PACs spanning the entire contig were scanned for chimerism using FISH and fiber-FISH, which detected no such event. The same experiment confirmed the order of these clones as shown in Figure 1 (data not shown).
Genomic Sequence of the 1q21–q22 Region between theCD1D and FCER1A Genes
To establish the nucleotide sequence around the CD1 region, two BACs (456N8 and 527I23) and seven PACs (810N20, 893N23, 987D5, 713I11, 974O18, 683M11, and 622B6), which collectively span a 1.1-Mb segment between the CD1D and FCER1A genes, were subjected to shotgun sequencing (Figs. 1, 2A). The 1,139,684 bp-long sequence (accession nos. AB045357–AB045365) was determined with a high redundancy of over 7. Overlaps between all BAC/PAC clones were ascertained at the sequence level. The overall G + C of the sequence is of 38.4%, which corresponds to the relatively A + T-rich isochore L1 (Fig. 2F; Bernardi 1995). This G + C content is, however, much lower than the densely packed 6p HLA region, which belongs to the G + C-rich isochore H1 (46.2%) (Fukagawa et al. 1995; Tenzen et al. 1997; The MHC Sequencing Consortium 1999). A closer inspection of the G + C content reveals fairly uniform dispersion throughout the entire segment, although numerous high G + C content peaks (>50%) were locally detected, and in most cases associated with expressed genes and/or CpG islands, including recognition sites for rare CG cleavage enzymes (Fig. 2E). When this 1.1-Mb region was scanned in 100-kb intervals, two 200-kb segments (the first linking the CD1D to CD1E loci and the second around the FY gene; physically located between 900 kb and 1,100 kb in Fig. 2A) at each end of a central 700-kb cluster were found to contain higher than average G + C contents, for instance, 40.0% and 41.3%, respectively. In contrast, the central 700-kb segment spanning nucleotide positions 200 kb to 900 kb in Figure2A is comparatively G + C-poor, with 37.1% on average (35.8% to 38.5%) (Fig. 2F).

Structural feature of the 1.1-Mb (1,139,684 bp) region from theCD1D gene to the FCER1A gene. (A) An operational contig constructed by an overlapping set of two BAC (456N8 and 527I23; in boxes) and nine PAC clones (810N20, 893N23, 987D5, 713I11, 974O18, 683M11, and 622B6) was subjected to nucleotide sequencing. (B) Gene map. Pink boxes indicate previously mapped genes. Red boxes depict genes newly mapped in this study. Green boxes show possibly expressed sequences. Black boxes refer to pseudogenes. Upper boxes define genes oriented from centromere to telomere (from left to right), whereas lower boxes show the opposite orientation. (C) Location of di-, tri-, tetra-, and penta-nucleotide microsatellite repeats. (D) Plot of the local G + C content in overlapping 200-bp windows. A red line indicates the average G + C content (38.4%). (E) Recognition sites of the restriction enzymes, NotI,BssHII, EagI, and SacI. (F) Plot of the local Alu and LINE repeat contents in overlapping 100-kb windows. Red and blue lines represent Alu andLINE repeats, respectively.
Analysis of the complete sequence with the RepeatMasker2program unveiled the following numbers of repeats: 332 Alus, 191 MIRs, 367 LINEs (LINE1+LINE2), 133 LTRs, and 23 MERs. These repeats collectively occupy 47.4% of the sequence, with Alus and LINEs representing 4.5% and 30.8% respectively, which corresponds to a density of one repeat per 3.4 kb and 3.1 kb, respectively (Fig. 2C).LINE1 comprises 28.0% of the LINE sequences. A 300-kb segment between the CD1D and CD1E genes as well as a 200-kb segment around the MNDA gene (nucleotide positions 600–800 kb in Figure 2A) reveal high LINE1densities, 40.7% and 35.6%, respectively. In contrast, a 100-kb segment around the SPTA1 gene (nucleotide position 500–600 kb in Figure 2A) displays low LINE1 density of 11.1% (Fig. 2F). Finally, a total of 406 microsatellites, 70 di-, 79 tri-, 156 tetra-, and 101 penta-nucleotide repeats (Fig. 2D), were also identified within the sequenced 1.1-Mb region (one repeat per every 2.8 kb), very similar to the frequency observed in the HLA class I region (Shiina et al. 1999).
Gene Content
The 1.1-Mb genomic sequence stretching from CD1D toFCERIA was subjected to gene identification analysis usingBLAST, GRAIL, and Genscan. This analysis revealed the existence of 41 genes within this segment or one gene per every 27.8 kb. These loci include two novel expressed genes (AIM2 andBL1A), 10 known expressed genes (CD1D, CD1A, CD1C, CD1B, CD1E, SPTA1, MNDA, IFI16, FY, and FCER1A from centromere to telomere), 12 possibly expressed sequences (ELL2-hom and 11OR), and finally, 17 new pseudogenes (nine OR, andRPS10, KIAA0696, RB1, HMG14, kinectin, PIG8, HSPCAL1, HRAD1) (Fig. 2B; Table 2). Focusing on the frequency of expressed genes (12), one gene per every 94.9 kb, one notes a gene density comparable to that (one gene every 70–100 kb) observed upon sequencing of the entire chromosome 21 (Ewing and Green 2000; Hattori et al. 2000) which, in turn, is almost identical to that assigned to the A + T-rich isochore L1 (Bernardi 1995). Most striking, among the 24 protein coding genes or possibly expressed sequences detected in this region, 12 are likely to fulfill immunological functions, highly redolent of the 6p-HLA region. These include CD1D, CD1A, CD1C, CD1B, CD1E, SPTA1, MNDA, IFI-16, AIM2, FY, BL1A, and FCERIA (Figs. 1, 2B).
Genes Identified around the CD1 Region
Genomic Architecture of the CD1 Region
The gene order of five CD1 genes, was established asCD1D—CD1A—CD1C—CD1B—CD1Efrom the centromeric side, spanning ∼176 kb (Fig. 2B; Table 2). The transcriptional orientation of CD1D, CD1A, CD1C, andCD1E was from centromere to telomere, whereas that ofCD1B was the opposite. These results support those previously published by Calabi and Milstein (1986), as well as Yu and Milstein (1989). Each of the CD1A–CD1E loci has been known to have two alleles, designated 1 and 2 (Han et al. 1999). The exonic sequences ofCD1A–CD1E, as established here, were in complete agreement with those from the human CD1 cDNA sequences (GenBank accession nos. M28825, CD1A; M28826, CD1B; M28827,CD1C; J04142, CD1D; and X14975, CD1E) and were found to correspond to allele 1 for all these loci. Moreover, all donor/acceptor splicing sites (GT/AG) including those of CD1E, which has 12 alternative splicing forms (EMBL accession nos.AJ289111–AJ289122), were of canonical nature.
It has been suggested that the present-day gene organization of theCD1 gene cluster was the result of regional duplication events from an ancestral CD1 gene (Calabi et al. 1989; Porcelli 1995;Porcelli and Modlin 1999), as similarly predicted for the HLA class I region (Shiina et al. 1999). To detect a possible trace for such regional duplication at the nucleotide level, the 200-kb CD1 cluster was subjected to dot-matrix analysis. However, in sharp contrast to the presence of multiple reiterated building blocks with remarkable contiguous homologies in the HLA class I region (Shiina et al. 1999), no evidence for any such internal duplication events could be obtained (data not shown).
A Novel Cluster of Olfactory Receptor Genes
Twenty new olfactory receptor genes (OR1-101–120 from centromere to telomere) were identified within our 1.1-Mb genomic sequence through an in silico search against various DNA databases usingFASTA and BLAST (Fig. 2B; Table 2). There was no rule governing the transcriptional orientation of these olfactory receptor (OR) genes. TheOR1-101–119 loci were composed of only one exon, whereas OR1-120 consisted of two (Table 2). Nineteen (OR1-101– -119) of these 20 loci were clustered within a 500-kb segment between the CD1E andMNDA genes. Interestingly, this cluster is not composed solely of OR genes (other OR clusters elsewhere in the genome tend to be pure of “intruders”) as it harbors three other loci, PIG8, HSPCAL1, and SPTA1 (Fig. 2B). The finalOR locus, OR1-120, was expelled to the end of the contig, 4.5-kb telomeric to the FCER1A gene. OurOR genes were classified into three different groups according to their structural characterization. Four loci (OR1-110, -113, -116, and -118) were classified as “fragmentary type” because of their short gene size (<300 bp), five (OR-103, -106, -107, -112, and -119) as “defective type” due to premature termination codons despite spanning a 906–939-bp stretch. Finally, the remaining 11 (OR1-101, -102, -104, -105, -108, -109, -111, -114, -115, -117, and -120) are intact, and therefore, likely to be “expressed” or “candidate” genes, despite the fact that no corresponding ESTs could be found in the current databases (this, however, might be expected, given the exquisite expression pattern of OR genes within the olfactory sensory neurons) (Fig. 2B; Table 2). Individual genes here are 924–975 bp in size, and carry seven transmembrane domains, common to all members of the G protein-coupled receptor (GPCR) gene superfamily (Mombaerts 1997).
To investigate the genetic relationship of these newly identifiedOR genes to other human olfactory receptor genes, a phylogenetic tree was constructed using the neighbor-joining method (Saitou and Nei 1987). The program was fed with nucleotide sequences extracted from the conserved, transmembrane segments 2–7, of all our sequences except for the fragmentary types, combined with those retrieved from 140 representative human OR genes deposited in GenBank and EMBL. As constructed, this phylogenetic tree allows theseOR genes to be classified into five major families corresponding to the previously classified families (G1, G2A, G2B, G3A, and G3B) as defined by Rouquier and colleagues (1998), based on percent nucleic sequence identity (NSI) of 87 OR sequences (Fig.3). All OR genes identified on the 1q21–q22 region belong to the G3B family indicated in blue in Figure 3. Interestingly, because ORs of the 1q21–q22 region except for OR1-119 were more closely related to each other than to any other olfactory receptor genes including the 7q33–35-located 669B10.3 gene (accession no.AC004853), they may represent new subfamilies of the G3B familily. On the other hand, OR1-119 was more closely related to the OR genes located on Chr.5, Chr.6, Chr.7, Chr.14, and Chr.17 than to the other OR1 genes. More importantly, all 1q21–q22 OR gene family members were more closely related to their OR counterparts encoded within the 6p21.3–22.2 region (indicated by yellow in Fig. 3) than to the OR genes in other families, and the branches found in the G3B family are longer than almost OR genes of other families. These findings suggest thatOR genes in the 1q21–q22 and 6p21.3–22.2 regions were created during the two rounds of duplication that generated the paralogous 1q21–q25 CD1 and 6p21.3–22.2 HLA regions (Kasahara 1999).
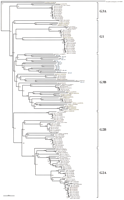
Phylogenetic tree of the olfactory receptor gene family. This phylogenetic tree was constructed employing the neighbor-joining method (Saitou and Nei 1987). Sequences were derived from the conserved region between transmembrane segments 2 and 7 in 156 olfactory receptor genes (five “defective type,”, 11 “expressed gene or gene candidate type” OR genes in 1q21–q22 and 140 human olfactory receptor genes submitted to GenBank). Five major families classified by this phylogenetic tree were designated G1, G2A, G2B, G3A, and G3B according to Rouquier et al. (1998). Blue and yellow boxes indicate the olfactory receptor genes located on 1q21–q22 and 6p21.3, respectively. Purple and orange boxes indicate olfactory receptor genes located on chromosomes 1 and 6, respectively, but within unknown subchromosomal locations.
Other Genes
Two other novel expressed genes were identified by sequence analysis of the 1.1-Mb region. The AIM2 gene (from nucleotide position 884696–899063) encodes an interferon-inducible protein (accession no.AF024714; DeYoung et al. 1997) and, interestingly, displays significant nucleotide homology to two neighboring centromeric genes. These areMNDA (from nucleotide position 656196–674270), which specifies a myeloid cell specific protein regulated by interferon α and IFI16 (from nucleotide position 832156–877358), which encodes the interferon γ-inducible protein 16 (Table 2). Indeed, exon 5 of AIM2 shares ∼60% nucleotide identity with exon 5 ofMNDA as well as exons 5 and 8 of the IFI16 gene. The other novel gene identified here is BL1A (from nucleotide position 993815–1025309). BL1A encodes a cell adhesion protein (accession no. F062733) with 43% amino acid similarity to the poliovirus receptor (accession no. P32506). Two isoforms have been reported for this gene, one containing exons 1–10, whereas the other consists of exons 5, 6, 8, 9, 10, and 11 (cDNA sequences BL1A, accession no. F062733, and FLJ10698, accession no. AK001560, respectively). SPTA1, known as one of the causative genes of pyropoikilocytosis, encodes an erythrocyte α-specific protein (Gallagher et al. 1991). This gene spans a 75.7-kb stretch, 254-kb telomeric to the CD1E gene (from nucleotide position 435886–511547), with a telomere-to-centromere transcriptional orientation. The exon/intron structure determined by comparison with the SPTA1 cDNA sequence (accession nos. J05244, M61852, andM61775–M61826; Sahr et al. 1990) was in complete agreement with that reported previously (Kotula et al. 1991). The gene is, indeed, sliced into 52 exons; all exon–intron boundaries were demarcated by canonical acceptor and donor splice sites except for the acceptor site of intron 32, which was GC instead of GT (data not shown).
DISCUSSION
To clarify the genomic structure of a critical piece of human chromosome 1q21–q22, one of four MHC-related paralogous regions, a dense 1.7-Mb YAC, BAC, and PAC contig linking the +P5 sequence to FCERIA was constructed. A 1.1-Mb internal subcontig, defined by eight BAC or PAC clones harboring CD1D andFCER1A at the centromeric and telomeric ends, respectively, was subjected to DNA sequencing determination and gene mining. Among the 41 genes identified, 27 were found to have paralogous partners on the other three regions (chromosomes 6p21.3–22.2, 9q33–q34, and 19p13.1–p13.4). These were CD1A–CD1E at 1q21–q22—HLA class I at 6p21.3–22.2, 20 OR at 1q21–q22—21 OR at 6p21.3–22.2, and SPTA1 at 1q21–q22—SPTAN1 at 9q34.13. These facts obviously support the previous prediction that this CD1 region on chromosome 1 was created by large-scale segmental duplications along with the other three paralogous regions (Katsanis et al. 1996; Endo et al. 1997;Hughes 1998; Kasahara 1999).
The presence of an OR gene cluster next to the CD1genes, paralleling the HLA region with an OR gene cluster on its telomeric side (Fan et al. 1996; Ehlers et al. 2000), is intriguing. Phylogenetic tree analysis allowed clear classification of these 20 OR genes into only one family, G3B (Fig. 3). Most of the OR genes in the chromosome 1q21–q22 region were probably created during the two large-scale duplications that resulted in generation of the paralogous CD1 1q21–q25 and HLA 6p21.3–22.2 regions (Kasahara 1999) given their greater genetic relatedness to paralogues located on 6p21.3–22.2 than to OR genes located within other families in our phylogenetic tree (Fig. 3). Finally, becauseOR genes are conserved from Drosophila melanogasterto human (AF156880; Parmentier et al. 1992; Ngai et al. 1993) and distributed over multiple locations in their genomes (Rouquier et al. 1998; Trask et al. 1998), structural and comparative analyses ofOR gene clusters from several species will make it possible to delineate the molecular dynamics of the evolutionary process through which the animal genomes evolved to the present-day complexity.
Another interesting feature of the 1.1-Mb CD1 region is that at least 23 of 37 expressed genes are immunologically relevant (Figs. 1, 2; Table 2). Again, this mirrors the HLA region, which contains at least 45 immune-affiliated genes among its 232 expressed genes (The MHC Sequencing Consortium 1999). This lends further support to the argument that the CD1 region is dedicated to the immune response, exemplified by CD1-mediated lipid or glycolipid presentation to various effector cells including γδ T, NKT, and NK cells (Porcelli and Modlin 1999) and, hence, is functionally equivalent to HLA-based peptide processing and presentation to αβ T cells (Davis and Bjorkman 1988). Based on this hypothesis, two models explaining the evolutionary generation of the MHC system can be proposed. One model is that two rounds of chromosomal duplication (chromosomes 1q21–q25, 6p21.3, 9q33–q34, and 19p13.1–p13.4) enabled two of the quadruplicate regions (chromosomes 1q21–q25 and 6p21.3) to be relaxed of functional constraints and thus allowed generation of the two major vertebrate (human) MHC systems (the CD1 and HLA-mediated antigen presentation systems). Another possibility is that the CD1 region represents the ancestral MHC system, which functioned as an “innate” immune system prior to the two rounds of chromosomal duplication (before the emergence of vertebrate). Two rounds of chromosomal duplication allowed one of the quadruplicate regions (chromosomes 6p21.3) to evolve into the HLA-mediated presentation pathway as part of the adaptive immunity system.
Dot-matrix analysis using the entire HLA class I region sequence (1.8 Mb) versus itself revealed numerous segmental duplications of a minimal building block,MIC—HCGIX—3.8-1—P5—HCGIV—HLA class I—HCGII (8–20 kb in size) (Shiina et al. 1999), whereas no such trace of duplication units was observed in the CD1 region. Within theHLA gene cluster, the occurrence of these repeated segmental duplications (which are the basis for the formation of the HLA backbone structure as well as a large variety of HLA class I genes) was estimated to have taken place some 20– 60 million years ago, as corroborated by dot-matrix and phylogenetic tree analyses (Shiina et al., unpubl.). Similar dot-matrix and phylogenetic analyses usingHLA class I gene sequences as well as two mouse and one ratCD1D sequences (Bradbury et al. 1988; Balk et al. 1991;Ichimiya et al. 1993, 1994; Kasai et al. 1997; Matsuura et al. 1997) indicates that the origin of human CD1 genes was some 60–100 million years ago, which places this event after the separation of mouse and human lineages (Porcelli and Modlin 1999; Shiina et al., unpubl.). Taken together, these findings suggest that the human CD1 region was established prior to the HLA class I region.
Of our 1.1-Mb sequence, 47% is composed of repetitive elements, among which the LINE1 sequences occupy the largest part, 28% (Fig.2F). This high LINE1 density, which corresponds to that of chromosome X (26%), is twice that observed in other autosomes (on average 13%) (Lyon 1998, 2000). Although no positive or negative correlation between LINE1 density and G + C content exists throughout chromosome X, fairly good positive correlation betweenLINE1 density and G + C content has been observed in most parts of the autosomes investigated, for example, chromosome 7 (Bailey et al. 2000). Generally, there is a positive correlation betweenLINE1 and gene densities along various segments of mammalian genomes (Smit 1999; Kazazian 2000). For instance, in our own previous experiment, the entire 1.8-Mb HLA class I region could be divided into five distinct segments based on nucleotide composition; within each segment a good positive correlation between LINE1 density and G + C content could be readily identified (Shiina et al., in prep.). In this context, it is notable that no significant positive correlation between the LINE1 density and the G + C content was observed in the 1.1-Mb sequenced region around the CD1 region (Figs. 2D,F). In this respect, despite being an autosome, this region of chromosome 1 may be more similar to chromosome X than to other autosomes such as chromosomes 6 and 7. Although the biological significance, if any, ofLINE1 elements remains unknown, it has been suggested that on the X chromosome, they act as a “booster stations” for a heterochromatinization signal transmitted by XIST RNA, which in turn, leads to X chromosome inactivation (Bailey et al. 2000). Therefore, it may be possible that this CD1 region undergoes autosomal imprinting or inactivation by unknown factors such as an XIST-like gene.
Furthermore, it is of great interest that susceptibility loci for a number of diseases such as elliptocytosis-2, spherocytosis, pyropoikilocytosis (Gallagher et al. 1992), autosomal dominant nonsyndromic deafness, autosomal dominant nonsyndromic sensorineural 7 (Fagerheim et al. 1996), familial hemiplegic migraine (Ducrons et al. 1997), familial partial lipodystrophy (Jackson et al. 1998), and familial schizophrenia (Brzustowicz et al. 2000) were mapped to the 1q21–q23 region. This region is also known for being involved in chromosomal translocations including those in certain lymphoblastic leukemias and papillary renal cell carcinoma, for example, t (1; 19) (q23; p13) and t (X; 1) (p11; q21), respectively (Williams et al. 1984;Weterman et al. 1996). Among the genes mapped here, SPTA1, which encodes a erythrocyte α-spectrin, has been well established as the causative locus for the development of elliptocytosis-2, spherocytosis and pyropoikilocytosis; 23 mutations have so far been detected in these patients (http://www.ncbi.nlm.nih.gov/htbinpost/Omim/dispmim? 182860). Moreover, the location of the familial schizophrenia gene was confined, by microsatellite-based mapping, to a 12-cM region on the telomeric side of +P5 (Brzustowicz et al. 2000), itself 200 kb centromeric of the CD1D gene. Approximately 20% of all reported cytogenetic anomalies seen in Wilms' tumor have involved chromosome 1q21–q22 (Slater and Mannens 1992). The 1.7-Mb YAC, BAC, and PAC contig around the CD1 region constructed in this study provides not only a powerful clue to dissect the binding site of several WT1 isoforms within the +P5 region but also a blueprint to carefully analyze 1q rearrangements occurring in Wilms' tumor. Indeed, some of the newly mapped genes, including the closely packed MNDA, IFI16 and AIM2 , which are of potential immunological relevance, may be actually involved in the development of Wilms' tumor, or some other cancer and/or mono-polygenic/complex disorder.
In summary, we have reported the genomic cloning and sequence analysis of a prototype MHC paralogous region on human chromosome 1q. The identification of a number of immunologically relevant genes and novel olfactory receptor loci lying in close vicinity to the MHC class ICD1 genes help to further define an emerging MHC-like functional cluster outside chromosome 6. This effort also eases positional cloning of disease-related mutations for a number of pathologies. In fine, similar high-resolution analysis on other segments of the human genome should help decipher the kinetics of vertebrate genome evolution in general.
METHODS
Construction of a YAC, BAC, and PAC Contig and Physical Mapping
Large insert yeast and bacterial clones were isolated by polymerase chain reaction (PCR)-based screening of the human CEPH (Centre d'études du Polymorphisme Humain) YAC library (Chumakov et al. 1992), a PAC library constructed from human lymphocyte DNA (Genome Systems Inc.), PAC and BAC libraries derived from human male lymphocyte DNA by Dr. Pieter J. de Jong (RPCI 4 and 5 series, and 11 series, respectively) (Osoegawa et al. 1996), and a BAC library constructed from the B cell line 978SK (Research Genetics). To construct a physical map of the 1q21–q22 region, 12 locus/gene-specific primer pairs were designed based on published sequences (Table 1), 18 STS primer pairs were selected from 70 markers positioned on the Whitehead Institute (http://carbon.wi.mit.edu: 8000/cgi-bin/contig/phys_map) WC1.16 contig map from WI-8369 (most centromeric) to UTR6608 (most telomeric), in the NCBI (http://www. ncbi.nlm.nih.gov/genemap) chromosome 1 Radiation hybrid map from D1S1600 (most centromeric) to D1S2635 (most telomeric), and 32 new STS primer sets that were prepared from 32 PAC and BAC end sequences. PCR analyses were performed using these PCR primers with YAC, PAC, and BAC DNAs as a template. PCR screening and physical mapping followed the protocol provided by Research Genetics andOsoegawa et al. (1996). Chromosomal mapping and chimerism of these BAC and PAC clones were checked by FISH, and the order of the clones within a contig was confirmed using fiberFISH as described previously (Takahashi et al. 1990, 1991; Mizuki et al. 1996; Suto et al. 1996). Southern hybridization analysis was carried out to confirm the integrity of the YAC, BAC, and PAC clones using PCR products amplified with locus/gene-specific primer pairs as probes (Inoko et al. 1986).
Sequencing Strategy
Two BACs and seven PACs covering the 1139-kb segment from theCD1D to FCER1A genes were shotgun sequenced (Deininger 1983; Wilson et al. 1994; Rowen et al. 1996). These cloned DNAs were purified by CsCl equilibrium density gradient centrifugation. Construction of shotgun libraries and preparation of sequencing templates has been described (Mizuki et al. 1997; Shiina et al. 1998, 1999).
DNA sequencing was performed by cycle sequencing employing AmpliTaq-DNA polymerase FS (PE Applied Biosystems), fluorescently labeled dye or BigDye primers, or dye or BigDye terminators in a GeneAmp PCR system (PE Applied Biosystems). A 373S or 377 ABI PRISM DNA sequencer was used for automated fluorescent sequencing (PE Applied Biosystems).
Assembly and Database Analyses
Individual sequences were minimally edited to remove vector sequences, transferred to a SPARC station (Sun Microsystems) on the TCP/IP protocol and assembled into contigs using theGENETYX-/SQ software (Software Development Co., Tokyo). Remaining gaps or areas of ambiguity were analyzed by a direct sequencing procedure employing PCR amplification products obtained with appropriate PCR primers or by nucleotide sequence determination of shotgun clones containing the segments of interest with sequencing primers designed from the sequence data and fluorescent dideoxynucleotide chain terminators (Wilson et al. 1994).
The final sequence was initially analyzed using GENETYXsoftware (Software Development Co.) on a Macintosh computer. Database searches (EMBL, GenBank, and DDBJ) were carried out using FASTA, BLASTN and BLASTX (Altschul et al. 1990). Because of the size limitation for sequence comparisons, dot-matrix analyses with varying parameters were used extensively to identify patterns of similarity. Searches for coding regions utilized theCRM/GRAIL, GRAIL I, GRAIL Ia, and GRAIL II gene-finding programs (http://avalon.epm.ornl.gov/Grail-1.3; Uberbacker and Mural 1991) and the GENSCAN gene-prediction program (http://gnomic.stanford.edu/∼chris/GENSCANW.html), along with the SwissProt database and the Smith-Waterman algorithm. Repeat and microsatellite sequences were detected with theRepeatMasker2(http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) andsputnik programs, respectively. Prediction of the transmembrane regions of ORctory receptor-like genes was determined using the SOSUI program (http://azusa.proteome.bio.tuat.ac.jp/sosui/).
Phylogenetic Analyses
Dot-matrix analyses were performed using Harrplot 2.0software (Software Development Co.). The phylogenetic tree was constructed employing the neighbor-joining method with sequences of the conserved region between transmembrane segments 2 and 7 of ORgenes (Saitou and Nei 1987). Multiple alignment of sequences and calculation of genetic distance were carried out using CLUSTALW(DDBJ; http://crick.genes.nig.ac.jp/homology/clustalw.shtml).
Acknowledgments
We thank Dr. Dominique Giorgi (CRBM, France) for providing us with olfactory receptor gene sequences. S.B. acknowledges support from the ACI Jeunes Chercheurs–Ministère de la Recherche and CReS-INSERM. Grants from the Japan Science and Technology Corporation (JST), an arm of the Science and Technology Agency, the Ministry of Education, Science, Sports and Culture, Japan, and the Tokai University School of Medicine supported this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵9 Corresponding author.
-
E-MAIL hinoko{at}is.icc.u-tokai.ac.jp; FAX 81 463 94 8884.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.175801.
-
- Received December 14, 2000.
- Accepted March 6, 2001.
- Cold Spring Harbor Laboratory Press








