
The Complete Human Olfactory Subgenome
- 1Department of Molecular Genetics and the Crown Human Genome Center, The Weizmann Institute of Science, Rehovot 76100, Israel; 2Bioinformatics Graduate Program, Boston University, Boston, Massachusetts 02215, USA; 3Department of Biological Regulation, The Weizmann Institute of Science, Rehovot 76100, Israel
Abstract
Olfactory receptors likely constitute the largest gene superfamily in the vertebrate genome. Here we present the nearly complete human olfactory subgenome elucidated by mining the genome draft with gene discovery algorithms. Over 900 olfactory receptor genes and pseudogenes (ORs) were identified, two-thirds of which were not annotated previously. The number of extrapolated ORs is in good agreement with previous theoretical predictions. The sequence of at least 63% of the ORs is disrupted by what appears to be a random process of pseudogene formation. ORs constitute 17 gene families, 4 of which contain more than 100 members each. “Fish-like” Class I ORs, previously considered a relic in higher tetrapods, constitute as much as 10% of the human repertoire, all in one large cluster on chromosome 11. Their lower pseudogene fraction suggests a functional significance. ORs are disposed on all human chromosomes except 20 and Y, and nearly 80% are found in clusters of 6–138 genes. A novel comparative cluster analysis was used to trace the evolutionary path that may have led to OR proliferation and diversification throughout the genome. The results of this analysis suggest the following genome expansion history: first, the generation of a “tetrapod-specific” Class II OR cluster on chromosome 11 by local duplication, then a single-step duplication of this cluster to chromosome 1, and finally an avalanche of duplication events out of chromosome 1 to most other chromosomes. The results of the data mining and characterization of ORs can be accessed at the Human Olfactory Receptor Data Exploratorium Web site (http://bioinfo.weizmann.ac.il/HORDE).
The vertebrate olfactory system can differentiate among millions of chemicals, which are detected by olfactory receptor (OR) proteins. These are encoded by the largest gene superfamily known to date, itself part of the G-protein coupled receptor (GPCR) hyperfamily. ORs were first characterized in rat (Buck and Axel 1991) a decade ago, and have been since detected in a large number of vertebrate species, including most orders of placental mammals, marsupials and monotremes, birds, amphibians, fish and lampreys (for review, see Mombaerts 1999; Glusman et al. 2000a).
To date, >300 human OR genes and pseudogenes have been reported (Parmentier et al. 1992; Selbie et al. 1992; Ben-Arie et al. 1994;Crowe et al. 1996; Vanderhaeghen et al. 1997; Buettner et al. 1998;Rouquier et al. 1998; Trask et al. 1998b; Bulger et al. 1999; Glusman et al. 2000a; Fuchs et al. 2001). Human ORs were frequently found to be clustered in the genome (Ben-Arie et al. 1994; Buettner et al. 1998;Rouquier et al. 1998; Trask et al. 1998a,b; Brand-Arpon et al. 1999;Bulger et al. 1999). The extents and locations of human OR clusters have been studied by fluorescence in situ hybridization (FISH) analysis (Trask et al. 1998a). This work demonstrated the presence of tens of human OR-containing genomic loci spread over most chromosomes, and suggested that the “olfactory subgenome” (the OR genes and their genomic environment) may include >0.1% of the human genome.
Detailed analyses of large-scale genomic sequences of human OR clusters provided the first direct understanding of the genomic structure of OR genes and of their organization into clusters (Glusman et al. 1996,2000b; Brand-Arpon et al. 1999), and a classification framework was provided (Glusman et al. 2000a). The evolution of OR genes was found to be profoundly influenced by the all-pervasive interspersed repetitive elements in their surroundings. These repeats can cause tandem gene duplications, can mobilize genes to more remote locations, and can even become part of their exon structure (Sosinsky et al. 2000).
By integrating such genomic information with the phylogenetic analysis of ORs, we could reconstruct the putative evolutionary history of the first completely sequenced OR gene cluster, on human chromosome 17 (Glusman et al. 2000b), apparently reaching back several hundred million years, into the amphibian stage.
The recently announced first draft of the human genome (International Human Genome Sequencing Consortium 2001) holds in it an unprecedented wealth of information, available for public study and scrutiny. We have now delved into this source, aiming to obtain a complete picture of the genomic structure and evolution of this large superfamily of genes.
RESULTS
The Human Olfactory Receptor Repertoire
We have performed a comprehensive data mining effort for OR genes in several data sources that together constitute the first draft of the human genome (International Human Genome Sequencing Consortium 2001). This led to the identification of a total of 906 human potential coding regions of OR genes and pseudogenes (hereafter referred to globally as ORs). These are located on almost all human chromosomes, with the exclusion of chromosomes 20 and Y (Fig.1).Two-thirds (601) of the ORs are newly detected, that is, not reported in previous publications, and the rest have been described elsewhere (Parmentier et al. 1992; Selbie et al. 1992; Ben-Arie et al. 1994; Carver et al. 1998; Rouquier et al. 1998;Trask et al. 1998b; Brand-Arpon et al. 1999; Glusman et al. 2000b). The 601 novel receptors described here had also not been annotated in GenBank, nor had they been included in previous classification work (Glusman et al. 2000a). The results of the data mining and characterization of ORs can be accessed at the Human Olfactory Receptor Data Exploratorium Web site (http://bioinfo.weizmann.ac.il/HORDE).
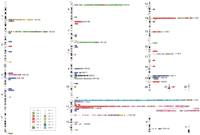
The human OR subgenome at a glance. ORs are depicted as squares, colored by family (see key). All Class I families are colored equally. Unclassified ORs are indicated in light gray. Framed squares denote intact genes. ORs to the left of each chromosome indicate singletons, and those to the right are in clusters of two or more. Gene order within each cluster is only approximate. The largest cluster on chromosome 11 is shown split for convenience. Small plus numbers under each chromosome name indicate the number of additional ORs for which a coordinate was not determined. Megabase coordinates (distance from the p telomere, excluding heterochromatic regions) were translated linearly into chromosomal localization. Therefore, the correspondence between cytogenetic bands and the indicated OR locations may be somewhat shifted.
Nearly 90% of the ORs were found in genomic sequences (Table1), half of which were confirmed by an additional independent sequence of any type (genomic, mRNA, etc). A significant majority of the ORs (681, 75%) spanned an interval corresponding to a full-length coding region (Fig.2a). Of these, at least 322 ORs had intact open reading frames, and are predicted to be functional. Data suggesting the presence of an expressed transcript are currently available for only a small fraction of these (<10%, Table 1), and from various tissue sources. The possibility of genomic contamination cannot be denied. On the other hand, when predicted functional OR genes are studied in detail (Sosinsky et al. 2000) most are observed to be indeed expressed.
Statistics of ORs Found in the Human Genome Draft

OR repertoire statistics. (A) Sequence-length cumulative distribution. The discontinuity near length 220 amino acids reflects the PCR product size obtained by using standard primers and is expected to disappear once the genome is finished. (B) Distribution of number of frame disruptions (frameshifts, in-frame stop codons, disrupting interspersed repeats, or partial coding regions flanked by non-OR genomic sequence). (C) Distribution of G + C content levels for the OR coding regions (CDS), their genomic environments (env), and the genome at large (genome), as subdivided into the three isochore groups L, H1–2, and H3, by using the 43% and 50% cutoffs. The 100% value refers to the 702 ORs for which an environment statistic could be calculated.
Isochore Distribution
In a gene cluster previously characterized by us, ORs were found to be located in a G + C poor, L isochore (Glusman et al. 2000b). Studying >5 kb windows surrounding OR coding regions, we now found this to be the general rule, with most ORs residing in a genomic environment corresponding to an L isochore (G + C < 43%, Fig.2c), a minority in H1–2 isochores (G +C of 43%–50%), and virtually none in the gene-rich H3 isochore (G + C > 50%) (Bernardi 1993). This distribution is biased compared with that of bulk human genomic sequences, indicating a preference for lower G + C contents, and in sharp contrast to the environment preferences of most genes (International Human Genome Sequencing Consortium 2001). In contrast, most of the OR coding regions have a G + C content in the intermediate range (43%–50%). On average, OR coding regions are 7.7% G + C richer than their environment, a trend shared by the exonic regions of most genes (Graur and Li 1999). The minority of ORs with G + C content lower than that of their surroundings are all pseudogenes.
Chromosomal Distribution
We have assigned approximate chromosomal megabase (Mb) coordinates to most OR-containing genomic clones by integrating information from the University of California at Santa Cruz's genome draft (http://genome.ucsc.edu) with mapping information from UDB, the Unified DataBase (Chalifa-Caspi et al. 1998) andhttp://bioinformatics.weizmann.ac.il/udb. This led to the assignment of chromosomal coordinates for 85% of the ORs (Fig. 1). Additionally, for 70 ORs a chromosomal assignment is available, but the Mb coordinate remains unknown. Only 72 ORs have no chromosomal assignment at present.
The chromosomal distribution of ORs is extremely biased, with six chromosomes (1, 6, 9, 11, 14, and 19) accounting for 73% of the repertoire. The remaining 27% are scattered on most other chromosomes, down to a single OR gene on chromosome 22 (Figs. 1,3a). Most strikingly, chromosome 11 alone has nearly half (42%) of all of the localized ORs. This observation, together with the unique genomic organization and diversity of ORs in this chromosome (see following) suggests a central role for chromosome 11 in the evolutionary history of the olfactory subgenome.

Distribution of ORs in chromosomes and clusters. (A) Number of ORs per chromosome (UL-unlocalized) sorted by decreasing OR numbers. (B) Distribution of cluster sizes. The thin line indicates the exponentially decaying expected cluster size, assuming a random chromosomal disposition. An expectation value of less than one is obtained for clusters of six ORs or more. The thick line indicates a power-law fit to the observed cluster distribution.
Olfactory Receptor Clusters
We analyzed the entire genome for the occurrence of OR clusters. The definition used was that two consecutive ORs >1 Mb apart belong to different clusters. The nomenclature proposed here for naming clusters is in the form of “chromosome@coordinate”, for example, “11@52” is the cluster on chromosome 11 at a position 52 Mb from the p telomere.
Simulation experiments indicate that if the genes were distributed randomly, no clusters would be expected to include more than five genes (Fig. 3b). In contrast, we observed 24 clusters with six ORs or more, and these clusters include 78% of the ORs for which a coordinate is available. Two clusters, both on chromosome 11, include more than 100 members each (Fig. 1). The number of ORs in a cluster appears to follow an inverse power-law distribution (Fig. 3b), in analogy to that demonstrated for compositional correlations in long DNA sequences (Bernaola-Galván et al. 1996). This suggests that OR gene clusters may have been enlarged by repeated events of local duplications of different lengths. Indeed, the internal structure of OR gene clusters shows subclustering by families (Fig. 1) and subfamilies (not shown).
The olfactory subgenome occupies nearly 1% of the human genome. The mean cluster size was ∼300 kb (excluding singletons), and 90% of the clusters had a size in the range 100 kb to 1 Mb. OR clusters were exhaustively searched for non-OR genes. Only the large clusters on chromosome 11 included long segments (up to 800-kb long) devoid of ORs but including other, non-OR genes. These may therefore be referred to as “super-clusters”. Excluding these non-OR segments, the two largest OR clusters (11@52 and 11@4) span 3.25 Mb and 1.4 Mb of sequence, respectively. By summing up the Mb spans of all observed clusters that belong to the OR subgenome, and assuming that singleton ORs occupy 10 kb each (likely an underestimate), the total amount of sequence occupied by all ORs (genome-wide) is computed to be ∼30 Mb.
Olfactory Receptor Pseudogenes
Of the 681 full-length ORs identified, 359 (53%) have one or more frame disruptions (frameshifts, in-frame stop codons, or disrupting interspersed repeats (Fig. 2b) and are considered to be pseudogenes. The pseudogene fraction is somewhat larger (63%) if partial sequences are included (i.e., those for which the full sequence will be available in the future). The fraction of pseudogenes observed in draft genomic sequences is higher than that from finished sequence, suggesting that the current results may somewhat overestimate the real pseudogene fraction.
We asked whether pseudogene formation tends to be a cluster-wide phenomenon. For this, an analysis was performed for each cluster, whereby the deviation from the genomic average pseudogene fraction was computed and a probability was calculated by assuming a binomial distribution. None of the clusters showed a significant deviation from the expected pseudogene composition, except for the 9@106 cluster, in which only 1 of 15 ORs is a pseudogene. It may be concluded that OR disruption is a random process targeted at individual genes.
The OR Sequence Space
We analyzed the identity score between each of the ORs and a representative data set of 55 non-OR GPCRs. The level of protein identity (PID) of ORs to their respective nearest GPCRs was 27.6% ± 2%, and none of the ORs showed more than 36% PID to any of the GPCRs studied (Fig. 4a). On the other hand, 96% of all ORs show >40% PID to their nearest neighbor. This suggests that the 40% cutoff efficiently discriminates between members of the OR superfamily and other GPCRs (Z-score for an OR to be 40% identical to a non-OR GPCR: 6.3). The relatively few OR sequences that show <40% identity to the most similar OR gene still show higher similarity to ORs than to non-OR GPCRs. Therefore, we assume that the OR data set is probably mostly free of contamination from irrelevant sequences.
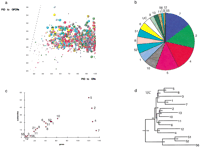
Phylogenetic classification of ORs. Color coding by family as in Figure1. (a) Protein identity score (PID) of each detected OR to its most similar OR and non-OR G-protein coupled receptor (GPCR). Spheres represent olfactory receptors; small spheres denote OR pseudogenes. Note the very different scales on both axes: The dotted line corresponds to x = y. (b) Relative sizes of the 17 OR families observed, and the unclassified ORs. (c) Correlation between the number of subfamilies and number of ORs in each family, averaging two ORs per subfamily, except for families 2, 4, and 7. (d) Phylogenetic tree of family consensus sequences. These were derived by using a majority rule at each position in a family-specific multiple alignment. The phylogenetic tree was derived by using the neighbor-joining algorithm in CLUSTALW with 1000 rounds of bootstrap. The percentage bootstrap strength is indicated at each node. The horizontal bar indicates 10% divergence along each branch.
We classified ORs into families and subfamilies on the basis of evolutionary divergence as published (Dayhoff 1976; Nebert et al. 1989;Lancet and Ben-Arie 1993; Glusman et al. 2000a). Only 35 sequences, all fragmentary pseudogenes, were left unclassified. We observed ORs belonging to the previously defined families 1–13 of Class II and families 51, 52, 55, and 56 of Class I. Four large families (2, 4, 5, and 7) include more than 100 members each, representing together 58% of the classified ORs (Fig. 4b). Six additional families (1, 6, 8, 10, 51, and 52) are of average size (30–70 members each), and seven are rather small (up to 25 members each).
We used principal components analysis (PCA) (de Leeuw 1988) to study the relationships among all of the ORs and the 55 GPCRs. This analysis is independent of the family classification and does not necessitate arbitrary cutoffs. PCA considers each gene as a point in multidimensional space, whereby the coordinates are provided by the similarity values of a gene to every other gene. PCA then finds a projection (a linear combination of dimensions) that best segregates the data. We calculated the principal components on the basis of intact ORs only. The initial PCA discriminates clearly between the outgroups and the two OR classes (Fig. 5a). An additional round of PCA, this time excluding the outgroups, clearly distinguishes between the Class I and Class II ORs (Fig. 5b). Such separation reflects the fact that, although Class II families are typically >35% mutually identical (average values for whole families), comparison across classes typically shows <30% mutual identity. The next round of sequential PCA (see Methods) separates family 4 from all other Class II families (Fig. 5c), suggesting that the generation of this family was an earlier evolutionary event. The remaining Class II families are segregated at higher PCA dimensions (Fig. 5d; PCA of individual subfamilies was not performed). When pseudogenes are plotted on the PCA, they are seen to generally be more diverged than the intact genes. The general trend is for pseudogenes to “drift” toward the origin of the coordinate system (Fig. 5). Similar results are obtained when calculating PCA coordinates on the basis of genes and pseudogenes together (data not shown).
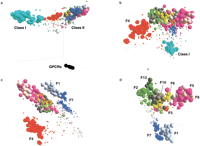
Visualization of the OR sequence space by sequential principal components analysis (PCA). Axes correspond to the first three principal components (linear combinations of the similarities of each gene with every other) and are shifted to be optimally visible: their intersection need not represent the cartesian origin (0,0,0). The vertical axis represents the first principal component. Spheres represent olfactory receptors; small spheres denote OR pseudogenes. The ORs are colored by family as in Figure 1. PCA shows the clustering of genes of common family. The first principal component, by maximizing the variation in the set accounted for, consequently separates the set into two groups (e.g., ORs and GPCRs in panel a). To further resolve the clustering of ORs, we remove the smaller group and run PCA on the remaining group. (a) PCA of all ORs including a diverse set of non-OR GPCRs. (b) PCA of all ORs. (c) PCA of Class II ORs. (d) PCA of Class II ORs excluding family 4.
Human Class I ORs Are Abundant and Probably Functional
Class I ORs have originally been identified in fish (Ngai et al. 1993) and subsequently found to be intermixed with Class II (mammalian-type) ORs in amphibian species (Freitag et al. 1995). Later, a small number of Class I pseudogenes was discovered in human (Buettner et al. 1998; Bulger et al. 1999; Feingold et al. 1999; Glusman et al. 2000a; Fuchs et al. 2001) but considered a minor evolutionary relic. Surprisingly, we have now detected a rather large number (102) of Class I ORs in the human genome, about one-tenth of the entire OR count, all located in the 11@4 super-cluster on 11p15. Even though this cluster includes much unfinished sequence, the pseudogene fraction among the ORs of Class I (52%) is considerably lower than that observed for Class II (77%). Expression data currently exists for six Class I ORs, in the form of mRNAs or ESTs.
Evolution of Sister Clusters
We have recently reported the analysis of a cluster (17@3) on chromosome 17 (Glusman et al. 2000b) that includes 17 ORs, mainly of family 1. A cluster of similar length and composition is now found on chromosome 9 (9@106; Fig. 6), which also contains mainly family 1 ORs. The two clusters have no subfamily in common, suggesting an early divergence. We found the subfamilies to be related in a pairwise fashion (Fig. 6) at 46%–55% PID. The analysis indicated the presence of seven subfamily pairs, and, accordingly, the occurrence of seven genes at the time of the duplication. Gene order and orientation were also somewhat conserved.
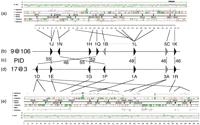
Sister-cluster evolution: Comparison between clusters 9@106 and 17@3. (a) GESTALT map of cluster 9@106, including graph of G + C content, GenScan predictions, interspersed repeats, and kilobase scale. Vertical gray lines indicate sequencing gaps. The order and orientation of the OR-containing contigs is final. Closed boxes represent intact ORs, and the open box indicates an OR pseudogene. The blue bar on top of the G + C content graph indicates an ∼10-kb segment surrounding OR5C1 , with G + C content corresponding to an H1 isochore. (b) Subfamily reconstruction of the 9@106 cluster at the postulated time of divergence from cluster 17@3. The dotted triangle indicates the single subfamily (1B) for which a counterpart was not observed. This subfamily is most similar to subfamily 1L, present in the same cluster, and may represent a gene duplication or deletion event that happened immediately after the cluster duplication. (c) Protein identity (PID) levels for each pair of subfamilies. (d,e) Subfamily reconstruction and GESTALT map of the 17@3 cluster, adapted from Glusman et al. (2000b), shown here in the opposite orientation. The recombinatorial events leading to the generation of OR1D4 and OR3A3 were omitted for clarity.
One of the surprising paralogous pairwise relations links the only ORs in these two clusters that do not belong to family 1. Thus,OR5C1, an OR of family 5 in the 9@106 cluster shows similarity to several ORs of family 3 in the 17@3 cluster. This is consistent with the phylogenetic tree observed for OR family consensi (Fig. 4d). It may be hypothesized that this cluster duplication marked the establishment of family 3, which evolved out of family 5, withOR5C1 representing their “evolutionary link”. The two families separate well in PCA (Fig. 5). Interestingly, the region surrounding OR5C1 shows a higher G + C content (Fig. 6), a relative rarity among ORs. Most likely, a family 5 OR from a G + C rich isochore invaded the 9@106 cluster before cluster duplication and was retained and expanded in the 17@3 cluster. This is in line with our previous observation that family 3 ORs have a higher G + C content than does the family 1 cluster surrounding them (Glusman et al. 2000b).
Global Cluster Evolution
To better understand the evolutionary pathways that led to the present human OR repertoire, we have performed a comprehensive comparison of the 24 clusters that contain six ORs or more. This was done by a novel, automated comparative cluster analysis (CCA), which formalizes the pairwise cluster comparison exemplified in the previous section. In brief, each pair of clusters was characterized by a metric that embodies the similarity of ORs within them (cluster identity level, CIL) and the probability that one of them arose from the other by partial or complete cluster duplication. Subsequently, a dendrogram was constructed on the basis of such pairwise comparisons (Fig.7). The results are consistent with two ancestral gene clusters, each containing solely members of one class: Class I on 11@4 and Class II on 11@52. The latter appears to have given rise to all other clusters by way of sequential cluster duplication, and it probably included at least one founder gene for each of families 1, 2, 4–6, and 8–10.
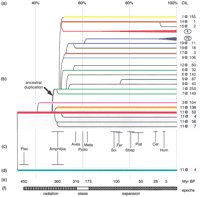
Comparative cluster analysis and OR molecular clock. (a) Cluster identity level (CIL) scale. (b) Dendrogram of duplicating clusters including Class II ORs. Clusters are named by using the “chromosome@Mb coordinate” nomenclature. Circled “4” and “7E” indicate the radiation of OR genes and clusters of family 4 (including clusters 9@71 and 3@136) and subfamily 7E (including clusters 6@186 and 19@63). Branches are colored according to the family (or families) that expanded most in each cluster (color key as in Fig. 1). A gray background highlights clusters on chromosome 11. The arrow indicates the ancestral duplication event that started the OR cluster radiation. 11@4represents here only the Class II section of the 11@4 cluster. (c) Taxonomical groups of the nonhuman species studied, including: Pisc(es): fish species; Amphibia: frogs, salamander; Aves: chick; Proto(theria): platypus; Meta(theria): koala; Sci(urognathi): marmot, mouse, rat; Fer(ungulata): pig, dolphin, dog; Strep(sirrhini): lemurs, squirrel monkeys; Plat(yrrhini): marmoset; Cer(copithecidae): baboon, macaques; Hom(inidae): chimpanzees, gorilla. Horizontal bars indicate the range of average PIDs when more than one species is included in the taxonomical group. (d) The single cluster containing Class I ORs. (e) Timescale in million years before present (Myr BP). (f) The three epochs of OR evolution.
This analysis also suggests that an early major event in the evolution of Class II ORs was the duplication of the almost complete ancestral Class II cluster, into what is now the q-arm sub-telomeric region of chromosome 1 (1@255; Figs. 1, 7), with a CIL of 47%. From this point in evolutionary history, the two clusters apparently had rather different fates: the original one (11@52) expanded within chromosome 11 by growing in size and by duplicating into new locations, including to the vicinity of the Class I cluster (11@6). In contrast, the new cluster (1@255) proceeded along the path of interchromosomal migration. It seems to have multiplied directly into at least six locations in the genome, and many of these propagated further into additional chromosomal neighborhoods.
Potential Orthologous Assignments
The assignment of orthologous pairs can be difficult for several reasons, including gene duplication events that occurred subsequent to speciation and unequal rates of evolution in different species and gene lineages. Such difficulty is compounded by the fact that usually the data sets are incomplete: the true ortholog of a gene in a given species may not have been observed yet.
We took advantage of having the human genome sequence with almost complete coverage to detect a most similar human gene (best hit, or BeT) for each nonhuman OR sequence detected in the present data mining effort. We then calculated for each represented species an average identity level for all of its BeTs (Table2). To avoid underestimates due to still-undetected human ORs and, conversely, overestimates due to contamination of nonhuman data sets with human sequence, we ignored BeTs with PIDs over 2 standard deviations away from the observed mean for each species. Translation into the million year (Myr) timescale for vertebrate evolution (Kumar and Hedges 1998) suggests a discontinuity in the evolutionary rate of the OR superfamily, between the mammal-bird and the eutherian-metatherian divergence times (Fig.7). The evolutionary history of ORs can thus be putatively divided into three major epochs of comparable lengths, which we call the epochs of radiation, stasis, and expansion (140, 137, and 173 Myr long, respectively). The average evolutionary rate of the intermediate epoch (3.6% per 100 Myr) is much lower than those of the first and last epochs (17.1% and 22% per 100 Myr, respectively). The typical CIL between duplicated OR clusters is 49%–56%, that is, after the establishment of the major families but before their subdivision into subfamilies (Fig. 7). This corresponds to the evolutionary period before the divergence from birds, suggesting that most mammals may harbor at least two dozen OR clusters.
Nonhuman ORs Detected
Subfamily-Specific Expansions
For most of the families, the average number of ORs per subfamily is surprisingly constant. This is manifested in a linear relationship between the number of genes and the number of subfamilies (Fig. 4c). The slope indicates an average of two ORs per subfamily, and a global calculation (including all families) shows an average of three ORs per subfamily, or over five ORs per subfamily if singleton subfamilies are excluded. Families that obey this “two ORs per subfamily” rule are likely to represent ancient divergence events, in which gene duplication took place for a certain period of time and then stopped. Thus, for a typical gene in such families there is only one other gene with an identity score higher than 60%. There are, however, three families that show significant deviation from the slope of 2. These are families 2, 4, and, to a much higher extent, 7 (Fig. 4c). The simplest interpretation is that certain subfamilies within such families have undergone a recent flurry of gene duplication and hence have many more ORs.
Most subfamilies (>85%) are chromosome- and cluster-specific. On the other hand, some specific OR subfamilies have undergone a striking scattering phenomenon. One subfamily of 7 (7E) dispersed to at least 35 genomic locations on almost all chromosomes (Fig. 1), in what is probably a primate-specific evolutionary trait. Likewise, some subfamilies of family 4 together expanded into over 15 locations throughout the genome.
DISCUSSION
The Structure of the Human OR Subgenome
The full characterization of the human olfactory subgenome is significant for a number of reasons. By identifying the number of functional human olfactory receptors, we have provided crucial information for understanding the genomic basis of combinatorial information encoding in this pathway (Lancet 1986; Kauer 1991; Lancet 1991; Mori and Shepherd 1994; Malnic et al. 1999). Of equal interest, considering the size of the OR family, the elucidation of the olfactory subgenome within the human genome provides an outstanding opportunity toward the evolutionary reconstruction of the OR superfamily. Such a molecular archeology may assist a general understanding of the expansion of gene families in the vertebrate genome.
Previous estimates of the size of the human OR repertoire have ranged widely, from a rough extrapolation of 130 ORs (Ben-Arie et al. 1994) to about 1000 on the basis of a molecular recognition model (Lancet et al. 1993). These estimates signify that the human repertoire is likely to be larger than the few dozen ORs expected in fish (Ngai et al. 1993) and comparable to that of rat (Buck and Axel 1991). However, so far, there had been no experimentally based estimate of the actual size of the human OR subgenome. By studying the draft of the human genome (<85% of the genome, excluding PHASE_0), we now observe >900 human ORs, of which ∼800 arise from studying large genomic sequences. This suggests that once the human genome is finished, the overall number may rise to 1000 or more, in very good agreement with the theoretical prediction (Lancet et al. 1993). ORs have been shown to be contained in large genomic segments, which have duplicated to many locations in the genome (Trask et al. 1998a), particularly near telomeres (Trask et al. 1998b). The presently reported OR repertoire size may therefore represent an underestimate, if significant numbers of almost identical OR loci are uncovered in the future. The OR subgenome constitutes ∼1% of the DNA length of the human genome, an order of magnitude larger than previous estimates (Trask et al. 1998a), but not out of line with what is indicated by the functional gene fraction, assuming that the human genome includes at least 35,000 functional genes (International Human Genome Sequencing Consortium 2001).
The present report provides a first global genomic map at <1-Mb resolution of the olfactory subgenome. This is made possible by the recent availability of large-scale human genomic sequence (International Human Genome Sequencing Consortium 2001) and of integrated megabase maps based on the human genome draft (Chalifa-Caspi et al. 1998 andhttp://bioinformatics.weizmann.ac.il/udb; the University of California at Santa Cruz's resource at http://genome.ucsc.edu). As a result of this analysis, it became possible to delineate a rather complete picture of the clustering pattern of ORs throughout the genome.
The overall localization of ORs on all chromosomes except 20 and Y is in agreement with previous work based on fluorescence in situ hybridization (FISH) (Rouquier et al. 1998). There is a rather good match also between the FISH-derived chromosomal locations of ORs and the results of the sequence data mining. On the other hand, several clusters and many lone ORs were now observed that have not been reported by the FISH analysis, such as the large centromeric OR cluster in chromosome 1. There are also cases of discrepancies in the location, for example, on chromosome X, where the FISH results show hybridization near the p telomere, whereas the present major localization is in the middle of the q arm.
A major outcome of the OR mapping results is the discovery of a disproportionately large OR count on chromosome 11. There were previous hints for OR-rich regions on this chromosome, suggesting seven distinct OR clusters (Buettner et al. 1998) and three of the strongest FISH signals (Rouquier et al. 1998). However, these previous results did not predict the fact that a full 42% of all human ORs are present on this single chromosome. Chromosome 11 is also unique in that it contains the most diverse collection of OR families: it is the only one that contains Class I receptors, and it has nine of the 13 Class II families. Interestingly, it also has the two largest OR clusters in the entire genome, each with >100 ORs. These findings provide convincing evidence that the genomic region presently represented by human chromosome 11 was the origin of the olfactory receptor repertoire. Notably, this chromosome shows contiguous conserved synteny in species dating back from human as far as the earliest mammals (O'Brien et al. 1999), even though it is now split into fragments in some species such as mouse.
The Genome-Wide Evolution of OR Genes
Our CCA provided a powerful tool to analyze the evolutionary history that has led to the present genome-wide disposition of ORs. It is a unique case in which a very large, well-defined vertebrate gene superfamily is subjected to a systematic formal scrutiny, by using the availability of the first nearly complete vertebrate genome. It appears that genome-wide expansion was initiated from chromosome 11 (Fig.8) but went indirectly through an early duplication to chromosome 1 (1@255). Interestingly, the timeframe for this initial cluster duplication is compatible with that of the second tetraploidization event of vertebrate evolution. Indeed, the q-telomeric region of chromosome 1 and the centromeric region of chromosome 11, where the ancestral OR clusters reside, have been shown to be paralogous since those ancient times (Jekely and Friedrich 1999).
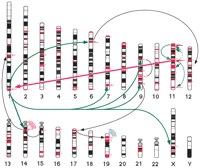
A tentative schematic view of the migration of OR genes in an “out of chromosome 11” scenario, based mainly on the cluster composition analysis (see text and Fig. 7 for details). The proposed steps, in rough chronological order, are (1) a duplication on chromosome 11 that resulted in the formation of the first class II cluster out of an original class I cluster (thick cyan); (2) a duplication that led to the formation of a cluster on chromosome 1, most likely in the framework of a whole genome diploidization (thick magenta); (3) internal duplications within chromosome 11 (thin green), and expansion from chromosome 1 onto a number of other chromosomes (thick green); (4) additional isolated duplications (thin black); gene scattering of family 4 (red “radio waves”) and family 7 (blue “radio waves”). Only the generation of clusters larger than five members is explicitly shown. The small red histograms to the right of the chromosomal bands indicate OR gene cluster locations, with the area proportional to cluster size (see http://bioinfo.weizmann.ac.il/HORDEfor details).
We hypothesize that the major driving force for the multichromosomal proliferation resides in the properties of this chromosome 1 cluster. It is not unlikely that two different mechanisms have been at work during the major steps in the radiation of OR clusters. The chromosome 11 repertoire expansion may have been the result of an intrachromosomal duplication mechanism, leading, among others, to the formation of two “super-clusters”. On other chromosomes, a second process that enabled the copying of ORs among different chromosomes has led to further repertoire augmentation.
Many of the inferred cluster duplications have very similar CIL values, lowering the confidence on the specific evolutionary pathway described in Figure 7. Nevertheless, the idiosyncratic gene contents of each cluster allows the reconstruction of several directional events of partial cluster duplication. The main potential pitfalls of this analysis include the assumption of nearly constant evolutionary rates on all clusters, and the possibility of gene conversion, which could lead to erroneous cluster lineage reconstruction. Although this has been demonstrated in the primate lineage (Sharon et al. 1999), such data are hard to obtain for events that occurred much earlier in vertebrate evolution.
Another important result of the CCA, coupled with the detection of potential orthologs for nonhuman ORs, is the delineation of the potential timeframe for OR cluster evolution. In the earliest stage, presumably before the emergence of amphibians (>400 Myr ago), precursors of most of the extant OR families appeared by local gene duplication. Next followed the radiation to multiple chromosomes, around the era of amphibians (300–400 Myr ago). Subsequently, a lengthy period of relative quiescence took place, lasting perhaps 150 Myr, with only minor further local duplication and diversification. This is manifested in the fact that most subfamilies are cluster-specific and in the significantly reduced evolutionary rate observed. Finally, in the last 10 Myr, the primate repertoire was subject to the combined effects of many functional genes turning into pseudogenes (Sharon et al. 1999; Gilad et al. 2000), and of extensive expansion of pseudogenes. Because human pseudogenes were allowed to be BeTs of nonhuman ORs, this could lead to some underestimation of the average PIDs and overestimation of their corresponding Myr values.
Class I ORs
The most ancient event that can be inferred in the evolution of ORs is the initial split between Class I (fish-like) and Class II (tetrapod-specific). In the clawed frog Xenopus these were shown to be expressed differentially in water- or air-accessible cavities, respectively (Freitag et al. 1995), but in the terrestrialRana, only expression of Class II ORs was observed (Freitag et al. 1998). Conversely, in the coelacanth Latimeria and in dolphins, both aquatic species, all observed Class II ORs were pseudogenes. This evidence and more (Freitag et al. 1998) suggested that Class II ORs are specialized for recognizing airborne odorants, whereas Class I ORs bind water soluble odorants, and hence that Class I ORs in mammals are evolutionary relics, now useless. It therefore comes as a surprise to find a large cluster of human Class I ORs, half of which are apparently functional, under selective pressure to maintain functional motifs. Expression of Class I ORs has already been reported in rat (Raming et al. 1998) and in human (Feingold et al. 1999). The rat Class I OR was shown to be expressed in a defined zone of the olfactory epithelium, suggesting that functional conservation may not be restricted to the OR coding sequence alone.
Class II families are all present in more than one chromosome each, except for the very small family 12. In sharp contrast, all human Class I ORs for which a coordinate could be ascertained are located in a single large cluster (11@4). Why did they not migrate to other chromosomes when interchromosomal duplications appear to be the rule? Most such duplications appear to have followed the invasion of chromosome 1, and OR gene duplications from chromosome 11 into other chromosomes appear to have been rather infrequent events. Therefore, Class I ORs may have remained clustered by chance alone. Alternatively, Class I expression might depend on the presence of regional control sequences, similar to the locus control region of β globin genes (Ho and Thein 2000). Because we could find no evidence for such a mechanism for Class II ORs, this would represent a severe functional difference between the two classes.
Principal Components Analysis of ORs
The complete depiction of the mutual relationships betweenn sequences requires their visualization in an (n−1)-dimensional space, clearly a preposterous proposition for studying several hundred ORs. At the other extreme, a phylogenetic reconstruction effectively reduces the dimensionality to one, with a huge concomitant loss of information. We have used the technique of principal components analysis (PCA) to simplify the problem to three dimensions, while retaining the maximal amount of information. As much as 47% of the OR sequence variability was maintained on such reduction and additional variability information was retrieved by performing sequential PCA. We expect that this methodology can be of much wider use in visualizing the internal structure of large gene families.
PCA results are not based on the family classification scheme used (Glusman et al. 2000a). Rewardingly, the results of both analyses are mutually consistent. We have observed that, when calculating the principal components based solely on apparently functional genes and then plotting pseudogenes by using the same coordinate system, pseudogenes tend to ‘drift away’ from their respective families toward the origin of the coordinate system. This is conceptually equivalent to highly mutated sequences appearing to be nearer to the root of a neighbor-joining phylogenetic tree. Such visualization could in principle be exploited to predict additional potential pseudogenes among apparently functional genes, that is, when a translatable frame is maintained but the sequence similarity has decreased significantly. Alternatively, it could point out the existence of genes that have evolved new functionalities and are under divergent selection pressures.
A potential difficulty of the PCA method is sensitivity to sampling biases. Because PCA aims to account for a maximum amount of the variability in the data, overrepresented families could appear to be more dissimilar from the rest of the ORs than they are in reality. This effect does not appear to cause any major distortions in the analysis of ORs presented here. For example, the non-OR GPCRs we used segregate from all ORs in the first principal component of the initial round of PCA, despite their small numbers. Similarly, the Class I ORs segregate first in the next round of PCA, even though they are a minority.
The Pseudogenization of the OR Repertoire
We have observed nearly 1000 ORs in the human genome (a microsmatic species), not unlike the number expected in macrosmatic species (e.g., rodent, canine). What brings about the suggested difference in odor perception capabilities between macrosmatic and microsmatic mammals? This is likely to be the result of the fact that only one-third of the human ORs appears to be functional, consistent with previous reports showing a large proportion of pseudogenes (Rouquier et al. 1998) and a recent decline in the functional fraction (Sharon et al. 1999; Gilad et al. 2000). The human repertoire not only shows a large fraction of genes that became pseudogenes, but also shows a large proportion of pseudogenes that arose as such, that is, those of subfamily 7E (Fuchs et al. 2001). Such extensive recent multiplication of pseudogenes may have reduced the functional OR fraction without reducing the actual intact OR count. Therefore, the initial primate OR repertoire may indeed have been smaller than that of rodents.
We found pseudogenes to be intermingled with apparently functional genes. The distribution of pseudogenes is consistent with a scenario in which genes have become pseudogenes at random, potentially because of reduced purifying selective pressure on the whole repertoire (Rouquier et al. 1998; Sharon et al. 1999; Gilad et al. 2000). This is supported by the roughly exponential decay in the number of frame disruptions (Fig. 2b). In contrast to the classical view of pseudogenes, in which they are formed as disrupted copies of functional genes (by simple duplication or by retroposition) (Wilde 1986), many OR pseudogenes lack an obviously identified functional counterpart and probably represent previously bona fide members of the functional repertoire, disrupted through mutation.
The observed OR clusters lack a clear internal structure as observed for homeobox (Deschamps et al. 1999), beta globin (Ho and Thein 2000), and immunoglobulin genes (Berek and Milstein 1988). Rather, OR genes appear to be disposed in haphazard arrangements that appear to correlate only with their phylogenetic classification. This, together with the large number of singleton OR genes scattered throughout the human genome, suggests that there may be no functional importance to clustering. ORs could remain clustered simply as a result of the process of expansion-modification (Bernaola-Galván et al. 1996), frequently mediated by interspersed repeats.
METHODS
Data Mining
A data mining pipeline was constructed to detect all available OR-like sequences in the public databases and to update the results as new database versions are released. TBLASTN (Altschul et al. 1997) was used to compare amino acid query sequences to the nonredundant version of GenBank (partitions nt, htg, and est_human, all updated to August 6, 2000), with a nonstringent expectation value cutoff of 1e-4. The queries used included 96 curated OR sequences representing all known families and 249 additional entries from the Human OR Data Exploratorium, HORDE (Glusman et al. 2000a), generated in many laboratories (Parmentier et al. 1992; Selbie et al. 1992; Ben-Arie et al. 1994; Crowe et al. 1996; Vanderhaeghen et al. 1997; Buettner et al. 1998; Rouquier et al. 1998; Trask et al. 1998b; Bulger et al. 1999;Glusman et al. 2000a; Fuchs et al. 2001). In a second round, 105 newly mined mouse genes and 344 newly mined human genes were used as additional queries (all data sets are available electronically,http://bioinformatics.weizmann.ac.il/papers/HORDE). All resulting database entries were cataloged by species and subdivided into four types: mRNA, EST, DNA, and genomic, the latter including entries annotated with keywords HTGS_PHASE1, HTGS_PHASE2, or HTGS_PHASE3, or with length at least 10 kb. Low-pass genomic sampling sequences were excluded (keyword HTGS_PHASE0). This yielded 908 human genomic clones, 351 additional human DNA entries (e.g., genomic PCR fragments), 379 human mRNAs, and 317 human ESTs. In addition, we used a set of 132 olfactory sequence tag (OST) sequences (Fuchs et al. 2001) and 21 EST assemblies obtained from the LEADS database (Compugen, Tel Aviv, Israel; http://www.cgen.com). Finished genomic sequences were analyzed as such, and unfinished sequences were parsed into contigs according to annotation or, where unavailable, according to runs of at least 50 Ns. The 18,386 contigs thus produced were analyzed for interspersed repeats by using RepeatMasker (Smit and Green 1997). Subcontigs were defined as segments between interspersed repeats, not including simple repeats and low-complexity regions.
Localization of ORs
Genomic localization was done on the basis of the July 17 freeze of the University of California at Santa Cruz's Working Draft Sequence (UCSC “GoldenPath”; http://genome.ucsc.edu), which presents a tentative assembly of the finished and draft human genomic sequence based on the Washington University-Saint Louis clone map (http://genome.wustl.edu/gsc). A coordinate was assigned to each finished or unfinished genomic clone, in megabases (Mb) from the p telomere of the given chromosome. In parallel, we used the Unified DataBase (UDB; http://bioinformatics.weizmann.ac.il/udb) to assign similar Mb coordinates to the clones, on the basis of their marker contents (Chalifa-Caspi et al. 1998). The two maps are largely colinear and were integrated on the basis of the coordinates of clones that could be localized in both. Clones for which no coordinate could be obtained by either method were provided only with a chromosome location according to UDB, by sequence similarity to another mapped clone, by annotation, or by e-PCR (Schuler 1997). The chromosomal coordinate of each OR was then calculated as the average of the coordinates of the genomic clones within which it is contained.
Detection and Classification of OR Sequences
Each subcontig was compared by using FASTY (Pearson et al. 1997) to a curated set of OR protein sequences from several species, yielding a conceptual translation product as described (Glusman et al. 2000a). We took into account the possibility of a pseudogene being disrupted by the insertion of interspersed repeats, with the two or more resulting parts being therefore located in different subcontigs. Such compatible candidate sequences were automatically joined into a reconstructed pseudogene. Whenever possible, all resulting sequences were trimmed or extended to use a suitable ATG codon for initiation and to end at a stop codon, avoiding such stop codons that would yield products shorter than 275 amino acids (no maximum limit was used). The sequences were finally split into OR or non-OR by comparing them to previously recognized OR sequences and to a representative, nonredundant database of 55 non-OR GPCRs extracted from Swiss-Prot (Bairoch and Apweiler 2000) with a 30% PID cutoff. To be automatically classified as an OR, a new sequence had to be at least 40% identical over at least 100 amino acids to another OR. A more stringent cutoff (50%) was required for sequences shorter than 100 amino acids.
A given gene could be represented in more than one overlapping sequenced clone. We removed such redundancy by considering two sequences as representing the same gene, if they are in the same chromosome, located in clones <300 kb apart and at least 99% identical at the nucleotide level. An exception to this rule occurred when two genes coappeared in the same clone, in which case they were considered to be distinct (only three such cases were encountered). It is possible that some very similar and neighboring genes could be misclassified as being the same, but we estimate this circumstance to be rare on the basis of our GESTALT analysis of complete clusters, which included >75% of all ORs detected. Sequences localized to a chromosome but without a coordinate were only compared to other sequences within that chromosome, and for those sequences lacking a chromosomal assignment the criterion of chromosome location was not applied.
For each resulting gene with more than one constituent sequence, a weighted consensus nucleotide sequence was created after multiple alignment by CLUSTALW (Higgins et al. 1996) by using the fast comparison parameter. The weighted consensus gave precedence to mRNA and genomic sequences over lower quality EST reads. This was followed by conceptual translation and end trimming to suitable start and stop codons, as described earlier.
ORs with a length of at least 275 amino acids without frame disruptions (frameshifts, in-frame stop codons, or disrupting interspersed repeats) were considered to be full length and apparently intact. Partial sequences without internal frame disruptions but disrupted by virtue of being embedded in non-OR sequence were defined as pseudogenes. Apparently intact ORs that were incompletely sequenced were excluded from the computations.
Each OR gene was assigned a family and subfamily by amino acid sequence similarity to previously classified OR genes, as described (Glusman et al. 2000a).
Isochore Analysis
To study the G + C content of an OR's environment, we used the unmasked sequences within 5, 10, 20, and 30 kb surrounding (but excluding) its coding sequence. The four resulting data sets yielded almost identical results (average difference in G + C content between sets was <1.5%). We therefore used the 5-kb environment range. Such G + C content values could be computed for 77% of ORs. Whole genome values were taken from the genome draft (International Human Genome Sequencing Consortium 2001).
Detection of Potential Orthologs
In addition to the human ORs, the data mining procedure yielded 851 OR sequences from 31 additional species. A BeT was determined for each of the nonhuman ORs detected as a result of the data mining effort, by comparing its conceptually translated sequence to the final set of human ORs and taking the hit with the highest PID. For each species, a divergence level from human was computed as the average PID of its ORs, excluding those more than two standard deviations away from the mean. A published molecular timescale (Kumar and Hedges 1998) was used to convert PID scores to million years before present (Myr BP).
Principal Components Analysis
A distance matrix was constructed representing the pairwise PID scores of ORs to each other and to 55 non-OR GPCRs. Each column in the matrix was normalized by dividing it by its standard deviation. Principal components were computed by using the Matlab package (MathWorks). Only apparently functional genes were used in this computation. The three first principal components were then used to map all genes, as well as pseudogenes, onto a three-dimensional space. Rendering of graphics and visualization were performed by usingSpotfire.net Desktop 5.0 (Spotfire Inc.,http://www.spotfire.com). In the sequential PCA method, clearly segregating groups of sequences are removed, and rounds of PCA are performed iteratively on the reduced data sets to visualize further sequence variability information.
Comparative Cluster Analysis
OR clusters were defined as the maximal groups of OR genes along one chromosome, such that the distance between two consecutive genes does not exceed 1 Mb. This cutoff was taken because of the low resolution of the mapping information used for assembling the genome draft. A tentative DNA sequence was built for each cluster by assembling all relevant finished and unfinished clones inSequencher (GeneCodes Corp). Although some uncertainty remains in contig orientation and order, this did not affect the analysis on the basis of the gene content of the clusters. To validate the correctness of the OR detection pipeline, we analyzed and visualized all cluster sequences by using the GESTALT Workbench (Glusman and Lancet 2000), including statistical analyses, gene prediction with GenScan (Burge and Karlin 1997), and recognition of interspersed repeats withRepeatMasker (Smit and Green 1997). This analysis revealed no additional ORs. Cluster sizes were calculated in kilobases directly from the reconstructed cluster sequences as the distance between the ORs at the ends of each cluster.
For each possible pair of OR clusters, we tested the hypothesis that one of them arose as a partial or full duplication of the other, and we determined the PID cutoff that best describes the divergence between the genes composing them. A cluster identity value IC was used in this analysis in the following way: For every ICvalue in the range 20%–100%, with an increment of 1%, the structure of both clusters was reconstructed by identifying later duplications. OR genes in each cluster showing a mutual identity of IC + 5% or more were defined as a “later duplication group” (LDG), potentially formed by local duplication after cluster divergence.
Subsequently, the clusters were subjected to a pairwise comparison among all possible pairs of LDGs. The identity value IGbetween two LDGs A and B (of different clusters) was defined as the average PIDs between all pairs of genes ai and bj, where gene ai is located in LDG A, and gene bj is located in LDG B. This analysis yielded a matrix of IG scores between LDGs of both clusters. We then identified those pairs of LDGs that represent mutual BeTs, that is, that show higher identity to each other than does each of them to the other LDGs.
These LDG pairs represent putative gene units at the time of cluster divergence, and their IG should be compatible with the postulated cluster identity level IC. This was tested by defining a score function f(IC as
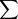 [5% − abs(IC − IG)] for every LDG pair for which abs(IC − IG) < 5%. A pair of clusters not sharing at least three such pairs was considered to be incompatible. Finally, the IC score that maximizes f(IC) was accepted as the mutual CIL. A parsimonious potential evolutionary history in the form of an OR cluster dendrogram was
then built on the basis of the gene contents of the OR clusters, as constrained by the estimated CIL values for each compatible
pair.
[5% − abs(IC − IG)] for every LDG pair for which abs(IC − IG) < 5%. A pair of clusters not sharing at least three such pairs was considered to be incompatible. Finally, the IC score that maximizes f(IC) was accepted as the mutual CIL. A parsimonious potential evolutionary history in the form of an OR cluster dendrogram was
then built on the basis of the gene contents of the OR clusters, as constrained by the estimated CIL values for each compatible
pair.
Acknowledgments
We thank Eitan Rubin and Compugen Ltd. for providing olfactory receptor sequence information from the LEADS database. D.L. holds the Ralph and Lois Silver Chair in Human Genetics. This work was supported by the Crown Human Genome Center, a Ministry of Science grant to the National Laboratory for Genome Infrastructure, the National Institutes of Health (DC00305), the Krupp foundation, the German-Israeli Foundation for scientific research and development and the Weizmann Institute Glasberg, Levy, Nathan Brunschwig, and Levine funds.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: The Institute for Systems Biology, 4225 Roosevelt Way NE, Seattle, WA 98105, USA.
-
↵5 Corresponding author.
-
E-MAIL doron.lancet{at}weizmann.ac.il; FAX 972-8-9344487.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.171001.
-
- Received November 13, 2000.
- Accepted March 13, 2001.
- Cold Spring Harbor Laboratory Press








