
Homogeneous Assays for Single-Nucleotide Polymorphism Typing Using AlphaScreen
Abstract
AlphaScreen technology allows the development of high-throughput homogeneous proximity assays. In these assays, signal is generated when 680 nm laser light irradiates a donor bead in close proximity to an acceptor bead. For the detection of nucleic acids, donor and acceptor beads are brought into proximity by two bridging probes that hybridize simultaneously to a common target and to the generic oligonucleotides attached covalently to the beads. This method allows the detection of as little as 10 amole of a single-stranded DNA target. The combination of AlphaScreen with allele-specific amplification (ASA) and allele-specific hybridization (ASH) has allowed the development of two homogenous single-nucleotide polymorphism (SNP) genotyping platforms. Both types of assay are very robust, routinely giving accurate genotyping results with < 2 ng of genomic DNA per genotype. An AlphaScreen validation study was performed for 12 SNPs by using ASA assays and seven SNPs by using ASH assays. More than 580 samples were genotyped with accuracy >99%. The two assays are remarkably simple, requiring no post-PCR manipulations. Genotyping has been performed successfully in 96- and 384-well formats with volumes as small as 2 μL, allowing a considerable reduction in the amount of reagents and genomic DNA necessary for genotyping. These results show that the AlphaScreen technology can be successfully adapted to high-throughput genotyping.
Single-nucleotide polymorphisms (SNPs), which are found every 250–350 bp, are responsible for most of the genetic variation that exists among human beings (Cargill et al. 1999; Hacia et al. 1999). Because of their high density, SNPs are viewed as invaluable tools for the mapping of genes implicated in complex human diseases, drug response, and drug metabolism (Schafer and Hawkins 1998; Zhao et al. 1998; Evans and Relling 1999; Kruglyak 1999; McCarthy and Hilfiker 2000; Roses 2000).
Numerous SNP genotyping platforms are being developed concurrent with the worldwide SNP discovery effort. Microchip technologies (Pastinen et al. 1997; Syvänen 1999; Chee et al. 1996; Wang et al. 1998;Winzeler et al. 1998; Fan et al. 2000; Pastinen et al. 2000) allow the parallel analysis of many SNPs but typically are used to analyze the SNP profile of a limited number of individuals. Several other technologies have been developed for, or adapted to, SNP genotyping, including mass spectrometry (e.g., Ross et al. 1998; Tang et al. 1999; Griffin and Smith 2000), pyrosequencing (Ahmadian et al. 2000; Alderborn et al. 2000), flow cytometry (Cai et al. 2000;Chen et al. 2000), fluorescence polarization (Gibson et al. 1997; Chen et al. 1999), cleavase assays (Lyamichev et al. 1999; Mein et al. 2000), and dynamic allele-specific hybridization (DASH; Howell et al. 1999). In general, these latter approaches are used to screen many samples for each SNP. In most genotyping assays, post-amplification steps such as purification of PCR products and additional enzymatic reactions are required. Multistep processes are costly in terms of reagent and labor and increase the risk of cross-contamination. Some approaches using fluorescence resonance energy transfer, such as TaqMan probes (Holland et al. 1991; Livak et al. 1995), Scorpion primers (Whitcombe et al. 1999), and Molecular Beacons (Tyagi and Kramer 1996;Tyagi et al. 1998; Marras et al. 1999), alleviate these problems by allowing the online reading of results, but they offer limited throughput and require expensive dual-labeled oligonucleotides (oligos).
AlphaScreen homogenous proximity assays were developed initially to measure interactions between biological binding partners (Ullman et al. 1994, 1996; Bossé et al. 2000). In these assays, a light signal is generated when a donor (D) bead and an acceptor (A) bead are brought into proximity (Ullman et al. 1994). The D beads contain phthalocyanine, a photosensitizer that generates short-lived singlet oxygen on irradiation at 680 nm. The singlet oxygen species diffuse only a short distance (∼200 nm) before decaying to the ground state. The A beads contain a mixture of chemiluminescer and fluorophores. On reacting with singlet oxygen, the chemiluminescer molecules undergo a series of chemical transformations that culminate in a time-delayed energy transfer to the fluorophores. The activated fluorophores, in turn, emit an amplified light signal at ∼600 nm, a shorter wavelength than the incident light. This cascade of reactions, coupled with time-resolved detection, results in high signal with very low background. The AlphaScreen technology has been developed successfully for high-throughput drug discovery applications (Bossé et al. 2000; BioSignal Packard Inc.) by using streptavidin-coated D beads and antibody-conjugated A beads.
AlphaScreen can be adapted for the detection of nucleic acids (Patel et al. 2000). For this purpose, generic oligos are covalently bound to the surface of the D and A beads. Currently, oligos of sequence dA24 are conjugated to the D bead and oligos of sequence d(AGTA)6 to the A bead, although other oligos could be used. To generate a signal, these two generic beads are brought into proximity by hybridization to two bridging detection oligos that can bind simultaneously to a common nucleic acid target (Fig.1). In this report, we show that the AlphaScreen technology is an exquisitely sensitive method for the quantitation of nucleic acids, with a detection limit in the low picomolar range. We also describe two high-throughput homogeneous genotyping methods based on the coupling of AlphaScreen detection with ASA or ASH. In both methods, PCR and probe hybridization are performed in the same reaction mix, and genotype analysis can be performed immediately after thermal cycling.
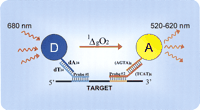
Schematic representation of the AlphaScreen detection of nucleic acids. See text for details.
RESULTS AND DISCUSSION
Sensitivity of AlphaScreen for the Detection of Nucleic Acids
The sensitivity and linearity of AlphaScreen was assessed in a probe assay (illustrated in Fig. 1), by using serial dilutions of a 47-mer single-stranded oligo target (Fig. 2A). Two bridging probes, containing sequences that are complementary to nonoverlapping portions of the target were used to bring the D and A beads to the oligo target. After probe and bead hybridization, the signal was read. A sensitivity of 10 amole was obtained, and the signal remained linear up to 3 fmole of target (Fig. 2A). An RNA target of identical sequence also could be detected with a similar sensitivity (data not shown). The specificity of the reaction was shown by the addition of genomic DNA to the detection mix. The AlphaScreen signal was not affected significantly by the inclusion of 1 ng/μL of denatured human genomic DNA (Fig. 2A), indicating that the assay is specific to the target and that AlphaScreen could be used to detect targets from complex mixtures.

Detection of nucleic acid targets. (A) Detection of a 47-mer oligonucleotide in the presence (diamonds) or absence (circles) of genomic DNA (1 ng/mL). (B) Detection of the BGT-1 PCR product after 20–34 PCR cycles. (open circles) Samples with 1 ng/μL DNA per reaction; (filled circles) no DNA controls.
The introduction of a denaturation step before probe hybridization allows the detection of double-stranded targets, permitting AlphaScreen to be coupled to PCR in a homogeneous assay in which probes and beads are included with the PCR mix during reaction setup. For this application, PCR annealing temperature should be above 63°C. High annealing temperatures avoid the potential interference of the bridging probes with the PCR. An example of the detection of a PCR product using AlphaScreen detection reagents is shown in Figure 2B. A region of the human Betaine/GABA Transporter 1 (BGT-1) gene was PCR-amplified and detected in a single-well reaction (Fig. 2B). Samples were removed from the thermal cycler every two cycles between 20 and 34 cycles and kept on ice. At the end of the assay, the samples were heated together at 98°C for 7 min to inactivate the Taq polymerase and denature the DNA strands. Detection then was performed as described in Methods. After only 20 cycles, a 6.5-fold signal-to-background ratio was obtained (Fig. 2B). Maximum signal was obtained after 30 cycles with 1,000,000 specific counts, which is a 500-fold signal to background ratio. At 32 and 34 cycles, there was a slight but reproducible decrease in signal due to excess target molecules that compete for probe binding and decrease the AlphaScreen signal. To avoid the occurrence of this effect, we determine the number of amplification cycles giving maximal signal for each assay by using a constant ratio of beads to probes (25 μg/mL of both beads and 10 nM of each detection probe). In most cases, equimolar concentrations of PCR product and probes will give a maximal AlphaScreen signal. In our experience, this usually occurs between 26 and 30 cycles of PCR.
Coupling of AlphaScreen to Allele-Specific Amplification
ASA has been used for several years for the genotyping of SNPs and small deletions/insertions (Newton et al. 1989; Okayama et al. 1989). The coupling of AlphaScreen to ASA has allowed the development of homogeneous assays amenable to high-throughput genotyping. Allele-specific PCR primers were designed with a second mismatch at the penultimate position of the primer to increase the specificity of the amplification reaction (Little 1995). AlphaScreen ASA assays were performed in two wells, one for each allele-specific primer pair. The reaction setup requires only the addition of the PCR/AlphaScreen mix (i.e., PCR reagents, bridging probes, and beads) and genomic DNA.
A validation study of AlphaScreen ASA was undertaken (Table1). First, a set of four assays was validated in-house for SNPs from the National Center for Biotechnology Information (NCBI) SNP database (http://www.ncbi.nlm.nih.gov/SNP/). The AlphaScreen results were compared to those obtained by direct sequencing of PCR products. A second set of assays was developed for two common SNPs found in the genes CDC45L and TBX1. These samples had been genotyped previously by single-stranded conformational polymorphism assay in the laboratory of one of the authors (M.L.B.) at the Division of Human Genetics, Department of Pediatrics, University of Pennsylvania. Five additional assays were developed for a set of WIAF SNP markers (Whitehead Institute SNP database; http://www.genome.wi.mit.edu/SNP/human/index.html). For these, validation was performed by comparing AlphaScreen ASA to TaqMan genotyping results obtained in the laboratory of Dr. Thomas J. Hudson (Montreal Genome Centre, McGill University, Montreal). A total of 355 correct genotypes were obtained from 358 samples, with accuracy > 99%. Two of the three discrepant samples were retyped with AlphaScreen and were confirmed to be heterozygous rather than homozygous, as originally determined. The third sample could not be reassayed because of lack of patient DNA. Amplification of an internal control allowing the detection of PCR failures would have detected at least two of the three miscalls found in the study. After these experiments, an internal standard has been incorporated into the assays (see below).
Validation of AlphaScreen Allele-Specific Amplification
A typical example of AlphaScreen ASA genotyping of 21 samples is illustrated in Figure 3. After allele-specific PCR and bead hybridization, the specific counts for each allele of SNP WIAF-896 were determined by transferring 10 μL of the reaction to a 384-well Proxiplate (Packard Instrument Co.) and recording the AlphaScreen signal. Raw data presented in Figure 3A show clear allele discrimination. Amplification from the allele A–specific primer gave slightly less signal than that of allele C, but in all wells in which amplification of allele A occurred, > 100,000 counts were detected.
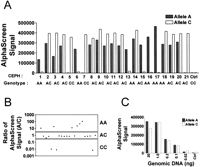
AlphaScreen ASA genotyping of SNP WIAF-896. (A) AlphaScreen signal obtained after the allele-specific amplification of WIAF-896 in 21 CEPH genomic DNA samples. (solid bars) Samples amplified with the A-specific PCR primer; (white bars) samples amplified with the C-specific primer. (Ctrl) No DNA sample. (B) Log plot of the allelic ratio of the AlphaScreen signal for the two alleles (A/C). Each point represents a single CEPH individual. (C) Titration of genomic DNA for the WIAF-896 AlphaScreen ASA assay by using an A/C heterozygote sample.
Figure 3B shows an automatically generated graphical view of the WIAF-896 genotyping results. The ratio of the counts obtained for both alleles was calculated and plotted on a logarithmic scale. Each point represents a sample. A/A homozygote samples had allelic ratios ranging from 10- to > 100-fold. Allelic ratios for the three C/C homozygotes were ≤0.01 indicating a 100-fold allelic discrimination. The A/C allelic ratios for the heterozygous samples were close to, but slightly lower than, 1.0 reflecting the lower signal observed for allele A in Figure 3A.
The minimum concentration of genomic DNA required for the AlphaScreen ASA assay was determined (Fig. 3C). The amount of genomic DNA in a 20-μL reaction was varied from 0.02 ng to 20 ng. This corresponds to a range of 6 to 6000 molecules of haploid genome per well. At 1 ng, a signal to background (S/B) ratio of > 30 was measured for both alleles of a heterozygous sample. At 0.1 ng, S/B ratios of between 8 and 16 were obtained. At lower concentrations of DNA, the signal approaches background levels. The extreme sensitivity of AlphaScreen detection should permit a major reduction in the amount of genomic DNA needed per SNP genotype.
Coupling of AlphaScreen to Allele-Specific Hybridization
A second high-throughput AlphaScreen genotyping platform, which combines AlphaScreen with ASH, has been validated. The AlphaScreen ASH offers the same design simplicity as AlphaScreen ASA: the PCR and AlphaScreen reagents are added to the wells as a single reaction mix with the genomic DNA. Separate wells are required for the detection of the two alleles. ASH detection is based on the hybridization of allele-specific bridging probes to the PCR product. This specific probe has the polymorphic base in the center of its target-specific portion and will hybridize preferentially to the perfect-matched allele, at a temperature that destabilizes the mismatched probe. Competing oligos complementary to the alternative allele, but lacking sequence complementary to the beads, are added to the PCR/AlphaScreen mix. These competing oligos have been found to increase the specificity of hybridization by the probes.
AlphaScreen ASH has been developed for seven SNP assays. The optimal hybridization temperature for allele discrimination was determined using a 96-well gradient thermal cycler in which every column was set to a different temperature (PTC-200; MJ Research). For the seven AlphaScreen ASH assays, the optimal hybridization temperature was found to be between 53°C and 63°C, primarily depending on the Tm of the allele-specific probe. The results obtained for the ASH genotyping of 21 Centre d'Etude du Polymorphisme Humain (CEPH) DNAs for SNP WIAF-896 assay are presented (Fig.4). In Figure 4A, the probe hybridization step is performed at 54.5°C, which is the optimal temperature for allele discrimination, although accurate genotyping results were obtained between 52°C and 56°C (data not shown). PCR could be performed using 28–33 cycles, without affecting the specificity of the assay (data not shown). The graphical view of the allelic ratio presented in Figure 4B clearly shows the sample genotypes. Titration of genomic DNA showed that the WIAF-896 ASH assay is extremely sensitive (Fig. 4C). Robust S/B ratios were detected using only 0.1 ng of genomic DNA per well, which corresponds to ∼300 copies of haploid genome. Decreasing the DNA concentration to 0.02 ng of genomic DNA still allowed the correct genotyping of the three possible genotypes (Fig. 4C; data not shown).
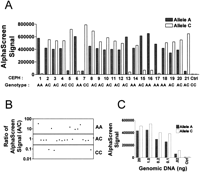
AlphaScreen ASH genotyping assay of SNP WIAF-896. (A) AlphaScreen signal obtained from 21 CEPH genomic DNA samples after allele-specific hybridization of the detection probes at 54.5°C. (solid bars) Samples amplified with the A-specific PCR primer; (white bars) samples amplified with the C-specific primer. (Ctrl) No DNA sample. (B) Log plot of the ratio of the AlphaScreen signal obtained for the two alleles (A/C). Each point represents a single CEPH individual. (C) Titration of genomic DNA for the WIAF-896 AlphaScreen ASH assay by using an A/C heterozygote sample.
As with AlphaScreen ASA, the accuracy of AlphaScreen ASH was determined. Five ASH assays were developed for SNP markers that had been tested previously with ASA assays (NCBI assay Id 394, 4568, 4621, 5173, and WIAF-896) and two assays for ApoE (C112R and R158C), a gene associated with late onset Alzheimer (LOA) disease (Corder et al. 1993; Poirier et al. 1993). All the genotypes obtained for the first five AlphaScreen ASH assays matched previous AlphaScreen ASA or direct sequencing results (Table 2). A set of 57 DNA samples, including 10 of known genotype from a LOA disease DNA panel (Coriell Cell Repositories), was genotyped for ApoE C112R and R158C mutations. All 114 ApoE genotypes from AlphaScreen ASH assays agreed with those of the LOA disease panel or with those obtained by direct sequencing of PCR products. An overall accuracy of 100% thus was obtained for ASH with a total of 225 correct genotypes.
Validation of AlphaScreen Allele-Specific Hybridization
Amplification Control
The most likely source of errors in genotyping in AlphaScreen will be from the failure of a sample to amplify. Thus, a heterozygote could be mistyped as a homozygote. To further improve genotyping accuracy, amplification of an internal control can be integrated to the AlphaScreen ASA and ASH assays to detect PCR failures. An additional PCR primer pair, which amplifies a control PCR product, can be included in the PCR/AlphaScreen mix. After the initial reading of the genotypes, probes specific for the control product are added to the samples, and a second hybridization step is performed. High AlphaScreen signal should be detected in every well in which amplification occurred. An example of the detection of a control PCR product for the WIAF-896 ASA genotyping assay is available as supplemental information athttp://www.genome.org.
A simpler approach has been developed for ASH, which avoids the addition of a second set of probes. Specific probe hybridization is first performed at the optimized hybridization temperature. An aliquot of the sample is read and the remaining sample is simply reheated, and the probes are reannealed at 37°C. At this lower temperature, most probes will show reduced discrimination for mismatched templates. A significant increase in signal should be observed for the mismatched allele in homozygous samples if amplification has occurred. An example of this approach with the ASH WIAF-896 assay is available as supplemental information at http://www.genome.org.
Genotyping from Complex Mixtures
Multiplex PCR procedures have been developed in several laboratories to save on reagents, time, and operating costs. Successful multiplex amplification of sets of 46 loci has been reported (Wang et al. 1998), but in most laboratories, the development of multiplex PCR is still a long process that allows, at most, multiplexing 8–10 different PCRs. As a preliminary demonstration of the potential of AlphaScreen for the detection of targets from multiplexed samples, PCR products for five ASH SNP assays were amplified separately and mixed after PCR. The hybridization of the allele-specific probes was performed both on the single samples and on an aliquot of the pooled samples. Correct genotypes were obtained for all the SNP markers in both the single and the pooled series, indicating that AlphaScreen could be used for the detection of multiplexed PCR products. The result of this experiment is represented graphically in supplemental information athttp://www.genome.org.
Automation and Miniaturization
To further automate the genotyping process and avoid cross-contamination of samples, we read AlphaScreen ASH reactions directly from sealed thermocycler plates (Fig.5). DNA samples of the three possible WIAF-896 genotypes were tested in a white 384-well thermocycler plate in 2-μL and 10-μL reaction volumes (Fig. 5A,B). After PCR and probe hybridization, the plate was read directly without removing the seal. The S/B ratio for homozygous samples was ∼10, and that of heterozygous samples was ∼1 independent of volume. There is a reduction in total signal observed with the 384-well PCR plate compared with the ProxiPlates due to the PCR plate geometry and reflectivity, and increased distance of the samples from the detector.
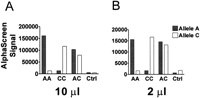
Miniaturization of the WIAF-896 AlphaScreen ASH assay. Samples representing the three genotypes were chosen. (A andB) Genotyping reactions in a 384-well format in which AlphaScreen signal was determined directly in the thermocycler plate. Reaction volume is indicated at the bottom of each panel. (Ctrl) No DNA sample.
Conclusion
In this report, we show that AlphaScreen can be used efficiently for detecting nucleic acids. The coupling of AlphaScreen to PCR has allowed the development of two novel homogeneous genotyping platforms. The AlphaScreen homogeneous genotyping assays have several qualities that make them attractive and competitive genotyping platforms. First, assays are straightforward to develop and simple to set up. Only two reagent additions are made to the PCR plates. The genotypes can be analyzed directly after PCR, as there is no need to remove free nucleotides, perform additional enzymatic reactions, or purify PCR products. Both types of assays are very robust. High allele discrimination is achieved (Figs. 3, 4), and genotyping results can be obtained from a wide range of DNA concentrations (Figs. 3C, 4C). The format of the AlphaScreen assays is flexible, ranging from single PCR tubes to a 384-well format, allowing easy scale-up and the automation of the entire genotyping process. AlphaScreen is compatible with multiplexing and assay miniaturization (Fig. 5). These features, combined with the fact that unlabelled oligos and generic beads are used for signal detection, will make AlphaScreen very cost effective for large-scale genotyping projects.
METHODS
Beads
AlphaScreen donor and acceptor beads are chemically modified latex particles of ∼250 nm in diameter. Oligonucleotides were coupled to the beads according to Patel et al. (2000). The binding capacity of the beads is measured as described (Patel et al. 2000). Approximately 7000 oligos are bound to each bead. AlphaScreen beads, loaded with generic oligos, are available from BioSignal Packard Inc. (www.biosignalpackard.com) for evaluation and purchase.
DNA Samples
Genomic DNA samples used for this study were obtained from various sources. In the AlphaScreen ASA validation study, samples for genotyping of the WIAF SNPs were from coded samples collected by the group of Dr. Thomas Hudson (Montreal Genome Centre, McGill University Health Center Research Institute, Montreal, Québec, Canada). The coded samples used for the genotyping of the TBX-1 andCDC45L were individuals recruited at the Division of Genetics, Department of Pediatrics of the University of Pennsylvania School of Medicine. For the other assays in the validation study, genomic DNA samples were obtained from the Coriell Cell Repositories. They included DNA samples from CEPH reference family members, from individuals in the DNA Polymorphism Discovery Resource, and from individuals in the late onset familial Alzheimer disease collection of the Aging Cell Repository of the National Institute of Aging.
Primer and Probe Selection
PCR primers of ∼30 nucleotides were selected. Pairs of detection probes, hybridizing to the same strand of a nucleic acid target, were designed with a target-specific region of 17 to 24 bases and with a 20-base generic tail. One of the probes had a dT tail complementary to the donor-bead oligos whereas the other had a d(TACT)5 tail, complementary to the acceptor bead oligos. In most cases, generic tails were at the 3′ end of the probe target-specific region to eliminate the possibility of DNA polymerization from the probe terminus. In some probes, the generic tail was added to the 5′ end. In this case, a 3′ phosphate group was added to prevent elongation from the probe terminus during PCR. Importantly, the pairs of bridging probes that were selected were unable to form stable heteroduplexes that could generate high background signal at room temperature. Primers and probes were purchased from Life Technologies. PCR primers were unpurified desalted oligos whereas bridging probes were purified with high pressure liquid chromatography.
Probe Assay
Probes (10 nM) and beads (25 μg/mL each) were mixed with serial dilutions of the target oligo in TKMB buffer (10 mM Tris-Cl, 50 mM KCl, 4 mM MgCl2, 200 μg/mL bovine serum albumin [BSA] at pH 8.0). After target denaturation for 5 min at 94°C, probes were hybridized for 5 min at 50°C and beads were annealed to the probes for 20 min at 37°C. A 10-μL volume of each sample then was transferred to a 384-well Proxiplate (Packard Instrument Company) and counted with the AlphaQuest-HTS (Packard Instrument Company). With this reader, samples are irradiated by a 680-nm laser for 0.3 sec before the recording of the light emission between 520 nm and 620 nm for 0.7 sec. Triplicate data points were made. The 47-mer target sequence was CTCCAATTTCTGACTGT GAGTTTTTGCGCTGCGAATAGAAAAAGTCT. The probe sequences were CTCACAGTCAGAAATTGGAGTGTTTTT TTTTTTTTTTTTTTT and TACTTACTTACT TACTTACTA GACTTTTTCTATTCGCAGCGC.
Detection of PCR Products
For the detection of BGT-1 PCR products, AlphaScreen beads and probes were added directly to the wells with the PCR components as a single addition. The PCR/AlphaScreen mix contains 250 nM of PCR primers, 10 nM of each detection probes, 25 ng/μL of donor and acceptor beads, 200 μM of TTP, dATP, dCTP, and dGTP, 0.01 U/μL of Taq polymerase (Qiagen), and 2.2 μg/mL of Taq-Start antibody (Sigma) in TKM buffer (TKMB without BSA). For the assay development data presented for the ASA and ASH WIAF-896 SNP assays, amplification was performed using 1 ng/μL of genomic DNA unless otherwise stated. For the validation of SNP assays using patient DNA, variable DNA concentrations were used, and some of the genotyping reactions were performed using < 0.1 ng/μL of patient genomic DNA. PCR conditions were set as follows: initial denaturation at 94°C for 5 min followed by 20–34 cycles of 10-sec denaturation at 94°C, 20-sec annealing at 68°C, and 30-sec elongation at 72°C. After PCR, the DNA polymerase was inactivated for 7 min at 98°C, and probes and beads were hybridized as described above. The PCR primers wereBGT-1 forward ACTCCAGGTCGGGGGCAGGGAAGCA CATGG andBGT-1 reverse GTGTGGGGAGGCAGCAGATGT TGGTGATGA. The probes for the detection of the amplified BGT-1 product were AGCTGACGGATTCTCTTACTTACT TACTTACTTACT and GCAGTGAGACTCTAGCTCTGTT TTTTTTTTTTTTTTTTTT.
AlphaScreen Allele-Specific Amplification Assays
For each AlphaScreen ASA genotyping assay, allele-specific PCR primers were designed with the polymorphic base at the 3′ position of the allele-specific PCR primers. A mismatch was introduced at the penultimate position of allele-specific primers to increase the specificity of the amplification reaction (Little 1995). The two alleles of each SNP were amplified in separate wells in the presence of beads and detection probes as described above for the probe assay. The PCR primers selected for the ASA WIAF-896 assay were forward 896-C-ASA, CCTTT GTCATCATAATCATATAGCCAAGGGACCC or forward 896-A-ASA, CCTTTGTCATCATAATCATATAGCCAAGGGACCAand reverse 896-ASA, GGCAGGGCACAAAACTGACATCA CATTGAAT. The probes for the detection of the amplified products were TGGAATGACTTGAAGCAGCCAAATACT TACTTACTTACTTACT and TTTTTTTTTTTTTTTTTTTTGCCA TCTCCCATTCTTTTCCTTC-PO4. Boldface letters represent polymorphic bases. The sequence of the primers used for the assays developed for the validation study is available on request. The sequence of the primers and probes used for the assays developed for the AlphaScreen ASA validation study are available on request.
AlphaScreen Allele-Specific Hybridization Assays
In AlphaScreen ASH assays, products encompassing the SNP were amplified in two separate wells by using the amplification conditions described above. In a first well, a probe with one of the SNP bases at the center of the target specific region was used together with a probe common to the two alleles. In the second well, the probe corresponding to the other SNP allele was used. The Tm of the common probe was equal to or higher than the Tm of the allele-specific probes. To mask the alternate allele product and increase specificity, we added 10 nM of a short oligo, matching perfectly the second allele, to the wells. After PCR, the samples were heated at 98°C for 7 min. The optimal temperature for allele-specific probe hybridization was determined using a gradient thermal cycler (PTC-200; MJ Research). Probe hybridization was performed at the optimal temperature for 5 min and was followed by the hybridization of the beads at 37°C for 20 min. The PCR primers used for the WIAF-896 assay were forward 896-ASH, CCCT TATCTTTTTAACCCCAAGACTTGTAG and reverse 896-ASH, TCCTGGCCTGCCTGAGAGGTTTTGGAATGA. Detection probes were probe 896-A, CCAAGGGACTAGGAATTTTGT TTTTTTTTTTTTTTTTTTT or probe 896-C, CCAAGGGACTCG GAATTTTGTTTTTTTTTTTTTTTTTTTT and common probe 896, CGATCATTTTCTCTGCCTATAGCTTACTTACTTAC TTACTTACT. The competing oligos were CCAAGGGAC TAGGAATTTTG and CCAAGGGACTCGGAATTTTG. Boldface letters represent polymorphic bases. The optimal probe hybridization temperature for the WIAF-896 was of 54.5°C. The sequence of the primers and probes used for the assays developed for the AlphaScreen ASH validation study are available on request.
Acknowledgments
We thank Dr. Thomas J. Hudson, Dr. Bing Ge, David Gagnon, and Joelle Collins for their participation in the AlphaScreen ASA validation study, Dr. Philippe Roby for bead labeling, Drs. Roger Bossé and Daniel Chelsky for helpful discussions, and Dr. Vera Webb for critically reading the manuscript. All human DNA samples were coded and processed according to approved protocols prior to our receipt of material. R.J.M. was the recipient of an industrial undergraduate research award from the Natural Sciences and Engineering Research Council of Canada.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL lbeaudet{at}biosignal.com; FAX (514) 937-0777.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.1725501.
-
- Received November 27, 2000.
- Accepted February 5, 2001.
- Cold Spring Harbor Laboratory Press








