
Using Mouse Genetics to Understand Infectious Disease Pathogenesis
The study of mouse and human genetic variation in infectious disease susceptibility (for review, see Malo and Skamene 1994; Hill 1998) should help to improve our knowledge of disease mechanisms by facilitating the identification of critical host proteins that modulate the infection process. Focusing on differences in disease susceptibility in humans will contribute to making progress in this field, but it is also possible to use mouse genetics to identify genes whose human orthologs are likely to affect the outcome of infections in man. It is impossible to adequately review all of the work that has been done to study the genetics of infectious disease susceptibility and pathogenesis. Therefore, I will discuss studies of two different bacterial infections. Although this is not a comprehensive approach, the examples help to illustrate my view that progress in understanding host susceptibility to infection will be facilitated by genetic studies of mouse models of infectious disease.
Susceptibility to Mycobacterial Infections
There are wide variations in susceptibility to mycobacterial disease in human and mouse populations (Bellamy 1998). Several mutations with profound effects on human mycobacterial susceptibility have been identified through the study of rare patients who presented with diseases caused by weakly pathogenic Mycobacterium spp. (Altare et al. 1998; Jouanguy et al. 1999a,b). In addition, it is likely that future genomewide screens in humans will solidify identification of novel genes involved in susceptibility to mycobacteria (Bellamy et al. 2000). However, mouse genetics offers a valuable complementary approach.
The study of mycobacterial pathogenesis and host defense has been powerfully influenced by the positional cloning of a mouse gene that affects susceptibility to diverse intracellular pathogens, including several (but not all) Mycobacterium spp., as well asSalmonella typhimurium and visceral Leishmaniasis pathogens (Vidal et al. 1993, 1995; North et al. 1999; Gruenheid and Gros 2000). This gene is currently called by the rather esoteric name of Solute Carrier Family 11, member 1 (Slc11a1), although the literature contains references to it by many other names, such as Bcg,Lsh, Ity, Nramp, and Nramp1. Slc11a1 is exclusively expressed in phagolysosomes within mononuclear phagocyte cells, where it acts to restrict growth of intracellular organisms (Vidal et al. 1993, 1996; Govoni et al. 1997;Gruenheid et al. 1997). By analogy to its mammalian homologSlc11a2 (also known as Nramp2, DMT1, andDCT1), Slc11a1 has been postulated to be a unidirectional transporter of divalent metal ions (Fleming et al. 1997;Gunshin et al. 1997). It may act by depleting the metal ion content of phagosomes containing intracellular pathogens, depriving them of iron, manganese, or other metals required for their proliferation. This model is consistent with the observation that wild-type Slc11a1appears to act by attenuating pathogen replication, rather than by promoting killing (Stach et al. 1984).
Amazingly, Slc11a1 is a member of a large family of metal ion-transporting protein genes that are contained in the genomes of organisms from bacteria to mammals (D'Souza et al. 1999; Curie et al. 2000; Kehres et al. 2000; Makui et al. 2000; Thomine et al. 2000). This fact raises the possibility that some intracellular pathogens and their host cells undergo a molecular struggle of orthologous proteins, with each attempting to gain an advantage through efficient sequestration of nutrients critical for the intracellular survival of the infecting microbe (for review, see Gruenheid and Gros 2000).
It is not yet clear whether Slc11a1 is involved in human resistance to mycobacterial pathogens. A few published case-control association and genetic linkage studies have provided evidence that the chromosomal region surrounding SLC11A1 influences human susceptibility to mycobacterial disease, although this influence is not observed in all populations (Shaw et al. 1997; Abel et al. 1998;Bellamy et al. 1998, 2000; Bellamy 2000; Greenwood et al. 2000). Although this needs to be investigated further, it is clear that the effect of SLC11A1, should there be one, is either strongly influenced by alleles of other susceptibility genes, or is somewhat weak and variable across populations. Nevertheless, despite the apparent infrequent occurrence of powerful functional polymorphism inSLC11A1, it is likely that its molecular action is identical to that of the mouse ortholog. As such, this gene (and its microbial homologs) is a viable starting point for research into the design of new therapeutic interventions to fight disease caused by intracellular pathogens.
Slc11a1 is not going to be the only mycobacteria susceptibility gene identified using mouse genetics. Recent work has identified two quantitative trait loci (QTL) on chromosomes 3 and 9 that have sex-specific effects on M. tuberculosisinfection-induced weight loss (Lavebratt et al. 1999). In addition, a locus on chromosome 1 (distal to Slc11a1) that influencesM. tuberculosis infection-induced mortality has been reported (Kramnik et al. 2000). The ultimate positional cloning of these mouse genes will undoubtedly provide other avenues of research into infectious disease mechanisms, as was seen in the case ofSlc11a1.
There are additional reasons to assert that studies in the mouse will contribute even more to the understanding of genetic differences in susceptibility to mycobacteria. The ability to make mutations in any mouse gene using homologous recombination offers the opportunity to test the involvement of specific host defense mechanisms in infectious diseases. Many mouse knockouts involving known immune system genes have been found to influence susceptibility to mycobacterial pathogens (e.g., see, Flynn et al. 1992, 1993, 1995; Cooper et al. 1993, 1997,2000; Dalton et al. 1993; MacMicking et al. 1997; Ehlers et al. 1999,2000; Sugawara et al. 1999). In some cases, these mouse models provide an opportunity to conduct detailed studies of mutations in mouse immune system pathways (Cooper et al. 1993, 1997; Dalton et al. 1993; Flynn et al. 1993) whose human counterparts are known to affect mycobacterial pathogenesis (for review, see Jouanguy et al. 1999a).
Susceptibility to Legionnaire's Disease
Legionella pneumophila is a significant community- and hospital-acquired pathogen that can cause either a severe pneumonia called Legionnaire's Disease or a milder febrile illness called Pontiac Fever (Bernstein and Locksley 1991). The estimates of the overall incidence of Legionnaire's Disease vary, but some studies suggest that Legionella infection may account for as many as 5%–10% of community-acquired pneumonia cases (Fang et al. 1990). The incidence of mildly symptomatic or asymptomatic Legionellainfection in human populations is unknown, although a few studies have suggested high rates of antibody positivity in certain populations (Sampson 1988; Bernstein and Locksley 1991). The reason for this apparent variability in human disease susceptibility is not known. Possible explanations include differences in the inoculation route or size, differences in the genetic background of the host, or genetic differences in pathogen virulence.
Legionella is a facultative intracellular parasite that exploits a poorly understood process within mammalian macrophage cells.Legionella's ability to cause disease is intimately linked to its ability to replicate inside of an unusual rough endoplasmic reticulum-bounded endocytic compartment (for review, see Ciancotto et al. 1989; Marra and Shuman 1992). Study of avirulent bacterial mutants has revealed that Legionella's intracellular survival is likely to be dependent on bacterial secretion of a nucleic acid or protein into the host cell via a type IV secretion apparatus (Winans et al. 1996). This macromolecular transfer, which occurs within minutes of the phagocytosis event, may alter the endocytic physiology of the host cell, preventing fusion of the Legionella-containing phagosome with lysosomes (for review, see Vogel and Isberg 1999).
Determining how Legionella exploits normal cellular processes during infection will be informative in terms of understanding disease caused by Legionella and in identifying macrophage defense functions that are also important in other infectious diseases. Fortunately, critical observations were made several years ago that have allowed the use of mouse genetics to study this problem. Cultured macrophages from different inbred mouse strains differ in their ability to support intracellular Legionella replication (Yamamoto et al. 1988, 1992). This difference in phenotype (in at least one permissive/nonpermissive strain combination) segregates in a Mendelian fashion (Yamamoto et al. 1991; Yoshida et al. 1991). Two groups independently have established that the gene responsible for the macrophage permissiveness difference (called Lgn1) maps to mouse chromosome 13 (Beckers et al. 1995; Dietrich et al. 1995).
Detailed molecular characterization of the mouse Lgn1 interval has demonstrated that it consists of a series of 80–100 kb direct repeats, each of which contains a paralogous member of theNaip gene family (Scharf et al. 1996) (Fig.1). The Naip genes, whose human orthologs (NAIPs) are also organized in a repetitive array, were described originally as candidate genes for spinal muscular atrophy (Roy et al. 1995). In addition, the Naip/NAIPgenes are members of a larger gene superfamily of BIR domain-containing proteins (BIRPs) (Miller 1999).
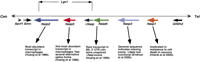
Map of the Naip gene family array in C57BL/6J. The horizontal line depicts the chromosome and the named arrows beneath the lines indicate the relative position and direction of transcription of the C57BL/6J mouse Naip gene paralogs. In addition, some flanking genes are indicated to provide genomic context.Naip6/ΔNaip gene encodes a 3′ UTR with unspliced exons from a nearby rearranged and nonfunctional ΔNaiplocus. The exact structure of the Naip gene family array varies among mouse strains. For example, mice of the 129 background have an expanded central portion of the Naip array that contains two additional close relatives of theNaip6/ΔNaip gene locus (Endrizzi et al. 2000;Growney and Dietrich 2000). Interestingly, the human orthologous region also contains repeats of NAIP gene sequences, although the map is uncertain because of considerable interhaplotype variability in structure (for review, see Growney et al. 2000). The genetic interval for Lgn1, which contains only Naip2, Naip5, and the 3′ UTR of Naip6/ΔNaip, is indicated above the chromosome. Arrows below each Naip symbol point to some relevant information about each gene.
The defining structural feature of the BIRP family is a zinc coordination motif that can mediate protein–protein interactions (the BIR domain) (Deveraux and Reed 1999; Miller 1999). Some members of the BIRP family can alter cellular susceptibility to apoptosis; this ability is dependent on the BIR domains (Deveraux and Reed 1999; Miller 1999). However, other members of the BIRP family appear to participate in chromosome condensation, alignment, and segregation during mitosis and meiosis (Speliotes et al. 2000).
Although some of the mouse and human Naip/NAIP proteins have been implicated in cell death pathways (Liston et al. 1996; Holcik et al. 2000; Mercer et al. 2000), it is not clear that this is their true physiologic role. Furthermore, the predicted proteins encoded by the mouse Naip genes are only 85% identical, with many nonconservative substitutions throughout, making it possible that the mouse Naip genes encode a diversity of functions (Huang et al. 1999). Recently, the critical interval for Lgn1 was narrowed to contain the entirety of Naip5 and portions of two otherNaip genes (Naip2 and Naip6) (Growney and Dietrich 2000). Because other transcriptionally active Naipparalogs exist outside this critical interval, it appears that the Naip proteins do not have entirely redundant molecular functions.
We will soon learn which of the Naip genes is responsible for the Lgn1 phenotype and whether structural variation in the human NAIP locus (Lefebvre et al. 1995; Roy et al. 1995; Growney et al. 2000) can influence susceptibility to Legionella. It will be important to determine whether Naip's effect onLegionella permissiveness involves a differential apoptotic response of the infected cell (Gao and Abu Kwaik 1999; Weinrauch and Zychlinsky 1999) or another, as yet unknown, Naip function.
In this vein, it is tempting to speculate that the Naip gene responsible for the Lgn1 phenotype may physically interact (via its BIR domain) with virulence factor proteins that are secreted by Legionella into the host cell. A recent report documenting increased expression of mouse Naip upon phagocytosis of bacteria or inert particles (Diez et al. 2000) can be viewed in light of a possible role for Naip in regulating endocytic traffic. If this hypothesis is true, Legionella may interfere with Naip activity to gain entry into a priveleged intracellular compartment.
Concluding Remarks
Even while investigators are swimming in genomic sequence information, the use of genetics to make unexpected connections between genes and phenotypes will remain a staple of gene function discovery. Because infections are a major cause of human morbidity and mortality (World Health Report 1999), I argue that susceptibility to infectious disease is among the most important phenotypes to study thoroughly. Certainly, a broader understanding of genes that influence host defense will translate into a greater likelihood of identifying cellular functions that can be manipulated for therapeutic goals.
Undoubtedly, human genetic methods will continue to impact our understanding of infectious disease susceptibility (Abel and Dessein 1998). In particular, the generation of high-density single nucleotide polymorphism (SNP) maps of the human genome may allow population-based searches for polymorphisms that are in linkage disequilibrium with disease status (Risch and Merikangas 1996; Kruglyak 1999; SNP Consortium). However, genetic analysis of phenotypic differences in mouse models of infectious disease represents a very strong complement to human genetic studies.
In this article, I have presented a few examples of how mouse genetics has begun to contribute to our understanding of infectious pathogenesis. Table 1 is a more comprehensive (though probably incomplete) synopsis of published mouse genetic studies of infectious disease that were not discussed in this article. A casual review of this table suggests that the genetic analysis of mouse models of infectious diseases will lead to the identification of several more infection susceptibility genes in the near future.
Mouse Genetic Models of Infectious Disease Susceptibility
Although it may be possible to design mutagenesis screens for infection phenotypes (de Angelis et al. 2000; Flaswinkel et al. 2000; Nolan et al. 2000), the data in Table 1 suggests that naturally occurring variation in mouse infectious disease susceptibility is already a very useful resource for finding important infection susceptibility genes. Historically, many pathogens have been studied in mice, and there is a large published body of data that may contain further information on genetic variation in susceptibility. Even in the absence of such clues, simple screens of existing mouse strains for variation in infectious disease-related phenotypes can be easily performed. In parallel, screens of inbred mouse strains that segregate the genomes of two parental inbred mouse strains [e.g., recombinant inbred strains, recombinant congenic strains, and chromosome substitution strains (Justice et al. 1992; Nadeau et al. 2000) offer a different way of uncovering genetic variation in disease susceptibility, even if the parental stocks show no difference in phenotype (P. Demant, pers. comm.). This is because disease susceptibility is often affected by alleles at multiple genes that can have complex epistatic interactions, and the segregation of parental genomes sometimes reveals the existence of genetic differences that cannot be appreciated through analysis of the parental strains.
The different evolutionary histories of mice and humans make genetic studies of mice particularly valuable as a complement to human genetic studies of susceptibility to infection. Mice are very likely to have numerous genetic polymorphisms in functions that do not commonly vary in human populations. By chance alone, an important host susceptibility gene could be polymorphic in mice, but rarely, if ever, polymorphic in humans. More ominously, there could be a lack of polymorphism in an important host susceptibility gene in either mouse or human because of the differential selective effects of the infectious diseases important in the history of the mouse and human populations. An illustration of the power of infectious disease as a selective force is provided by the high prevalence of apparently deleterious hemoglobin alleles among humans living in regions where malaria infection is extremely common (for review, see Weatherall et al. 1997). For these reasons, it is vital to scan for useful genetic polymorphisms in more than one mammalian host, to minimize the chance that a molecule that may be a key target of therapeutic intervention goes undiscovered.
Acknowledgments
I thank Rebecca Mosher, Victor Boyartchuk, James Watters, Fred Winston, Igor Kramnik, Jon Seidman, Susan Dymecki, Ralph Isberg, Nancy Andrews, and anonymous reviewers for their helpful suggestions and comments on various drafts of this manuscript.
Footnotes
-
↵1 E-MAIL ; FAX (617) 432-3993.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.173101.
- Cold Spring Harbor Laboratory Press








