
A cSNP Map and Database for Human Chromosome 21
- Samuel Deutsch1,2,
- Christian Iseli3,4,
- Philipp Bucher4,5,
- Stylianos E. Antonarakis1,7, and
- Hamish S. Scott1,6
- 1Division of Medical Genetics, University of Geneva Medical School, Geneva, Switzerland; 2Graduate Program in Molecular and Cellular Biology, University of Geneva Medical School, Geneva, Switzerland; 3Ludwig Institute for Cancer Research, Epalinges, Switzerland; 4Swiss Bioinformatics Institute, Epalinges, Switzerland; 5Swiss Institute for Experimental Cancer Research, Epalinges, Switzerland
Abstract
Single nucleotide polymorphisms (SNPs) are likely to contribute to the study of complex genetic diseases. The genomic sequence of human chromosome 21q was recently completed with 225 annotated genes, thus permitting efficient identification and precise mapping of potential cSNPs by bioinformatics approaches. Here we present a human chromosome 21 (HC21) cSNP database and the first chromosome-specific cSNP map. Potential cSNPs were generated using three approaches: (1) Alignment of the complete HC21 genomic sequence to cognate ESTs and mRNAs. Candidate cSNPs were automatically extracted using a novel program for context-dependent SNP identification that efficiently discriminates between true variation, poor quality sequencing, and paralogous gene alignments. (2) Multiple alignment of all known HC21 genes to all other human database entries. (3) Gene-targeted cSNP discovery. To date we have identified 377 cSNPs averaging ∼1 SNP per 1.5 kb of transcribed sequence, covering 65% of known genes in the chromosome. Validation of our bioinformatics approach was demonstrated by a confirmation rate of 78% for the predicted cSNPs, and in total 32% of the cSNPs in our database have been confirmed. The database is publicly available athttp://csnp.unige.ch or http://csnp.isb-sib.ch. These SNPs provide a tool to study the contribution of HC21 loci to complex diseases such as bipolar affective disorder and allele-specific contributions to Down syndrome phenotypes.
Single nucleotide polymorphisms (SNPs) (Kan and Dozy 1978) are likely to become widely used markers for the mapping of complex genetic traits. They are the most common type of genetic variation and, with rapidly developing technologies for genotyping, they are becoming highly suitable for automated, inexpensive, and high-throughput genetic analysis (Landegren et al. 1998; Brookes 1999).
Common sequence variants in or near genes are thought to be involved in the control of natural phenotypic variation, including the risk for common disorders, susceptibility to infection, and drug response (Lander 1996; Collins et al. 1997; Chakravarti 1999). This hypothesis, sometimes referred to as common disease–common variant (CD–CV), has generated considerable interest in SNPs over the last few years, resulting in concerted efforts for the characterization of large numbers of these sequence variants for genetic epidemiology studies.
It is expected that with large numbers of SNPs and cohorts of an appropriate sample size (Lander and Schork 1994; Risch and Merikangas 1996), positive associations between a phenotype of interest and the various interacting loci that influence it would be detected. However, recent experimental and theoretical studies (Harding et al. 1997; Clark et al. 1998; Lai et al. 1998; Kruglyak 1999; Moffatt et al. 2000) show that the linkage disequilibrium (LD) on which indirect association studies rely is not uniform throughout the genome, and might be limited in certain regions to intervals as short as 3 kb. In this case, ∼500,000 markers would be needed to cover the genome comprehensively (Kruglyak 1999). An alternative is to pursue a candidate gene approach in which genes of interest are carefully screened for SNPs, and these can then be directly tested for association.
SNPs can be generated experimentally in a number of ways (Underhill et al. 1997; Wang et al. 1998); alternatively, one can use the large amount of sequence data available in order to detect differences within clusters of high quality overlapping sequences. This has been proposed in a number of studies (Buetow et al. 1999; Garg et al. 1999; Marth et al. 1999; Picoult-Newberg et al. 1999), all of which use base-calling programs and traces available in the databases to discriminate between sequencing errors, paralogous alignments, and true polymorphisms.
We set out to identify a dense collection of cSNPs (single nucleotide polymorphisms on cDNAs) on human chromosome 21 (HC21). As part of this effort we developed a new algorithm for context-dependent SNP detection that generates high quality cSNPs without the use of trace data.
HC21 is the smallest human chromosome, and it is related to numerous monogenic and complex disorders (Antonarakis 1998). The entire nucleotide sequence of its long arm (33.6 Mb) has recently been determined with 127 known and 98 predicted genes (Hattori et al. 2000). We used this complete chromosome sequence as our template for EST alignment and SNP discovery, and we present the resulting variants in a web-based cSNP database and map (http://csnp.unige.ch/http://csnp.isb-sib.ch). We anticipate that this resource will be useful for the characterization of the various complex traits that map to HC21 and for the study of the complex phenotypes in Down syndrome.
RESULTS
Potential cSNPs were identified using two different strategies, a genome-driven approach and a gene-driven approach. For the genome-driven approach, the complete HC21 genomic sequence was extracted from public databases, filtered for repetitive and low complexity sequences, and used for searching human ESTs with a high level of homology. This approach obtained 25,701 ESTs, which were then clustered and aligned for automatic SNP discovery using a newly developed algorithm called snp_detect (see Methods). This program generates an output file consisting of multiple alignments of short genomic fragments containing the candidate regions that fulfilled our SNP criteria (Fig. 1).
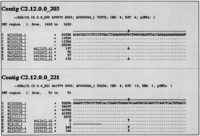
Example of snp_detect output. Precise mapping information of the alignment is given on the second line. C, R, and E denote genomic, mRNA, and EST sequences, respectively. Each sequence is hyperlinked to GenBank and to CGAP when traces are available. The first cSNP is contained in an anonymous EST, and the second in geneKIAA0539.
As an alternative method we performed a gene-driven cSNP search. We performed a batch BLAST search of 127 HC21 genes (comprising 379 kb of sequence), against all other human nucleotide databases, using PowerBLAST. Output files were then visually inspected for potential cSNP detection.
A third source of cSNPs was a laboratory-based gene-targeted approach, which was undertaken as part of an effort to identify pathogenic mutations in candidate genes for disorders that mapped to HC21 (Lalioti et al. 1997; Nagamine et al. 1997). We did not expect to detect all polymorphisms within these genes, since only a limited number of individuals were resequenced in each case; however, common variations were identified.
Using these combined strategies, 377 potential cSNPs were identified, as shown in Table 1. Of the 263 cSNPs that were automatically identified using the snp_detect algorithm, 69% were contained in known HC21 genes, and the rest represented SNPs in anonymous ESTs (98% correspond to predicted genes as reported by Hattori et al. 2000). All cSNPs were precisely positioned to genomic contigs and organized into a clickable map, as shown in Figure 2.
cSNP Identification

Example of cSNP map covering 1.1 Mb of sequence. Blue boxes represent genes placed on either side of the central axis according to the orientation of the transcript. Red lines indicate the presence of a candidate cSNP. Numbers represent megabases of sequence from centromere to telomere (as presented in Hattori et al. 2000).
cSNP Features
We analyzed the cSNPs in terms of the nucleotide substitutions observed and their effect at the amino acid level. The results shown in Table 2 are similar to previously published data (Garg et al. 1999).
cSNP Features
The 38 cSNPs predicted to have an effect at the protein level were further analyzed in terms of the nature of the amino acid substitution observed, using the PAM 250 comparison matrix. As expected, a high proportion of these changes were conservative (73%), confirming a strong effect of selection, because we estimated by simulation studies that random nucleotide substitutions would produce 49% of conservative changes.
Confirmations
To validate our data, we randomly chose 36 (10%) candidate variants for experimental confirmation and PCR-amplified the selected SNP regions using a pool of DNA from 10 individuals of unrelated CEPH families. The amplimers were then sequenced to determine the presence of polymorphisms at the expected positions (Fig.3).
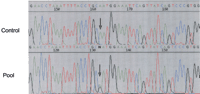
Example of cSNP confirmation in gene PWP2H. The polymorphic nucleotide is shown by an arrow.
In all cases single amplimers were obtained, and in 78% of the cases (28 of the 36) we could clearly observe the presence of the predicted variation. In all cases we sequenced the candidate variant region in single individuals to discard false positives caused by sequencing background.
Of the eight candidate cSNPs that could not be confirmed, one case resulted from bidirectional sequence failure and two others from sequencing of consistently poor quality, preventing accurate base calling. In two cases the predicted cSNPs were false positives caused by alignment of known paralogous sequences, whereas for the remaining three cases that could not be confirmed, no explanation was evident. These, however, are likely to be caused by unknown paralogous genes, or may even represent very rare polymorphisms.
Nucleotide Diversity
Sequence diversity can be expressed in a number of different ways, all of which describe the extent of polymorphism within a given sample. The simplest way to represent this is by dividing the number of polymorphic loci by the number of loci sampled (sometimes referred to as S value), but this is highly dependent on the number of chromosomes (n) assayed. Two estimates of variability that correct for sample size are commonly used, θ (the expected number of polymorphic sites for a given sample size) and π (the average heterozygosity per site) (Hartl and Clark 1997). Since the majority of the cSNPs were generated using bioinformatics methods, it is practically impossible to precisely calculate θ or π because the number of chromosomes sampled, n, is unknown. However, we can estimate this parameter if we consider that 95% of our alignments have an n that lies between 6 and 40, as determined by the number of different cDNA libraries from which the ESTs originated (assuming different cDNA libraries are derived from unrelated individuals). Variation within this interval has a relatively small impact on the value of θ, and hence we took the median as a reference for calculation (see Methods).
We thus estimated the value of θ to be 3.2 ± 0.7 × 10−4, using for this calculation the total cDNA length of known HC21 genes and the number of cSNPs found within these regions. This θ value (which under the infinite sites model is the same as π) is lower than those obtained using experiment-based approaches (θ = 5.30 × 10−4[Cargill et al. 1999] and θ = 8.0 × 10−4[Halushka et al. 1999]) but similar to other reports of bioinformatics-based SNP discovery (θ = 3 × 10−4 [Garg et al. 1999]).
To study the factors that might influence the SNP detection efficiency of our method, we plotted the average proportion of polymorphic loci (S value) for each gene against the number of different libraries present in the alignments (Fig.4). We obtained a correlation coefficient of 0.47, which is significant and indicates (as expected) that the number of independent ESTs in the alignment accounts for some of the variation in SNP discovery.
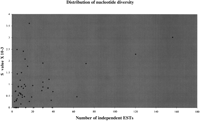
The graph shows the distribution of the observed nucleotide diversity against the number of independent ESTs. The correlation coefficient is 0.47. The S value corresponds to the number of cSNPs per nucleotide.
DISCUSSION
In this paper we present a dense, high quality cSNP map of human chromosome 21 containing on average 1 SNP per 1.5 kb of transcribed region studied, and covering 64% of all known genes in the chromosome. As part of this study, we developed a new algorithm for automatic cSNP detection, in which genomic sequences are used as a template for the construction of EST alignments (Marth et al. 1999) that can be screened for high quality mismatches. Unlike other approaches, thesnp_detect algorithm does not rely on trace data for quality discrimination. However, it incorporates a number of complex features for quality control (see Methods) that take into consideration the surrounding sequence as well as the quality of the alignment, the advantage being that a greater number of ESTs can be incorporated into the analysis, potentially providing a higher sensitivity for cSNP detection. In addition, EST traces are hyperlinked when available, and hence can be easily viewed by the user as necessary.
The high confirmation rate of cSNPs generated in this manner indicates that the context-dependent analysis (post-alignment treatment) performed by the snp_detect algorithm, in combination with a final step of visual correction (in which we eliminated a few potential cSNPs in which the presence of noncognate sequences was detected) was efficient at limiting the number of false positives in the database.
We estimated the level of nucleotide diversity for all known HC21 genes using standard population genetics approaches (Hartl and Clark 1997) and obtained a value of θ = 3.2 ± 0.7 × 10−4. This value is two- to threefold lower than those of similar diversity studies on different subsets of genes using more direct screening approaches such as solid phase resequencing and DHPLC (Cargill et al. 1999; Halushka et al. 1999), but similar to values obtained when using bioinformatics approaches (Garg et al. 1999).
We hypothesize that this underestimation of θ is owing to three main factors: (1) Rare variants or variants present in genes with low levels of expression (for which few ESTs are available) are likely to be missed. (2) The distribution of ESTs is significantly skewed toward the 3′ end of genes, hence cSNPs located near the 5′ end will be identified less efficiently. (3) The algorithms were designed to be conservative so that only a proportion of the observed variation was considered.
To test the first hypothesis we studied the relationship between nucleotide diversity among genes and EST coverage (only ESTs originating from different libraries were considered for this analysis). We observed large differences in the detected nucleotide diversity between genes (see Fig. 4) and found that an important fraction of the variability was caused by their level of expression (which directly relates to the numbers of independent ESTs available). Therefore, in poorly expressed genes (or in genes with a rare expression pattern) most of the nucleotide variation is likely to be missed. However, EST depth accounts for only part of the variance (because the correlation coefficient was relatively low), and other factors are likely to be involved. These include differences in selective constraints between genes, the nature of the sequence, the presence of recombination hot spots, pseudogenes, and Alu/LINE elements (Nachman et al. 1998; Conley et al. 1999; Fahsold et al. 2000). In addition, we observed that two-thirds of the cSNPs were located primarily in the 3′-UTR regions, whereas with direct screening approaches the distribution is generally uniform (Cargill et al. 1999). This indicates that, indeed, some of the 5′ variation in genes is being missed.
Several SNP databases are publicly available, and these include experimental as well as bioinformatics data. However, although they contain large numbers of variants, in none of these is there a systematic scan for cSNPs in genomic regions, which is the strategy that we describe here. We analyzed the overlap between our dataset and other databases and found it to be very small (Table3). This was particularly evident with the SNP Consortium (TSC) collection (see Table3), in which 2630 SNPs in human chromosome 21 have been experimentally identified. This small overlap can be explained by the fact that the TSC collection was generated randomly and does not target coding regions.
Overlap with Other Databases
We estimate by BLAST analysis that ∼60/2630 of the TSC chromosome 21 SNPs lie in transcribed regions, and this reveals one limitation of bioinformatics approaches, in that only ∼1/3 (24/60) of the variation that is likely to be present is actually being identified in our study (see Table 3). These numbers are consistent with our estimates of θ being two to three times lower than the values obtained from experimental approaches.
Because most relevant variation related to disease susceptibility is likely to be clustered in and around genes and cSNPs are still poorly represented in the public databases (as shown by the overlap data), a cost-effective way (Roberts 2000) to generate cSNP maps might be to use bioinformatics approaches such as the one presented here. However, one must consider that the effectiveness of indirect association studies has not been proven empirically and direct candidate gene-based association studies will probably require more comprehensive cSNP discovery approaches. This is being currently pursued for genes that are suspected to be involved in phenotypes of interest (Cambien et al. 1999; Cargill et al. 1999; Halushka et al. 1999; Ohnishi et al. 2000;Yamada et al. 2000).
The database presented here is the first extensive chromosome-specific cSNP collection, which is likely to contribute to the dissection of complex phenotypes that map to human chromosome 21.
METHODS
Sequence Handling, Clustering, and Alignments
From the EMBL database we extracted all sequences annotated as human chromosome 21 that were at least 10 kb in length using the getchrom script. These sequences were filtered against Repbase and Repsim (a database of simple repeats) to mask repeats and low complexity sequences.
All human mRNA and EST sequences were extracted from EMBL and used to perform high similarity matches against the filtered HC21 sequence subset produced in the first step using BLAST. As a result, we generated a table of genomic DNAs associated with their matching mRNAs and ESTs.
We then constructed clusters within the HC21 DNA sequences to group together overlapping and consecutive sequences. (During the final stage of the work we used the entire chromosome sequence in five contigs as released in Hattori et al. [2000].) For this we computed the best overall alignment for each pair of sequences and then distributed the pairs into clusters with the purpose of building contigs. Global alignments were performed using a specially designed script, blalign, so that the relative positions of particular sequences within the cluster (mapping) were deduced. We then partitioned each cluster in overlapping segments of 2000 bp. These segments were scanned, and all mRNA and EST sequences that mapped to these regions were integrated to generate multiple alignments.
SNP Detection Method
Candidate SNP regions were detected with the aid of a novel program called snp_detect (source code of the program is available fromftp://www.isrec.isb-sib.ch/sib-isrec/snp_detect).This algorithm scans a multiple alignment composed of genomic, mRNA, and EST sequences that are believed to originate from the same gene for the presence of sequence variations.
The sequence input has to be in FastaAlign format with specifically formatted header lines that assign to each sequence a corresponding sequence type (chromosomal, RNA, or EST).
The algorithm proceeds in three phases that can be defined as data preprocessing, SNP scanning, and postprocessing. The first operation of the preprocessing phase, which we term Gap treatment, classifies the gaps in the multiple alignment into short and long gaps, according to a length threshold. Short gaps are believed to represent sequencing errors or short indel polymorphisms (insertion/deletions), whereas long gaps correspond to introns or real indels. Two different gap characters are used in the internal representation to distinguish between these two cases in further processing steps. Terminal gaps are always considered long gaps, regardless of their actual length. The long gaps are automatically extended by a few positions in order to make the procedure more tolerant to alignment errors.
The gap-treated multiple sequence alignments are subsequently translated from single-base to k-tuple (oligonucleotide) representation. Different k-tuples are encoded by integer numbers between 0 and (4k − 1) in a standard fashion. The negative integer −1 is used as place holder for missing sequence information. There is a simple, although not immediately obvious, rule guiding this process: If a character in the single-base alignment is the last element of a contiguous string consisting only of kunambiguous nucleotide symbols (A, C, G, or T) plus a variable number of short-gap characters, it is replaced by the correspondingk-tuple code; otherwise it is translated into −1. Note that long-gap characters are treated like ambiguous base characters and thus cause the production of −1, whereas short-gap characters typically lead to duplication of the same k-tuple at adjacent positions.
The k-tuple conversion functions as an input data quality filter via at least three mechanisms: (1) by masking (replacement by −1) bases in the vicinity of ambiguous base calls, (2) by ignoring possibly mispositioned gap characters within otherwise identical oligonucleotides, and (3) by requiring potential SNP alleles to occur several times in the same context (see below).
During the SNP detection phase, each column of the k-tuple alignment is subjected to the following SNP test: If at least two different k-tuples are observed at frequencies equal to or greater than both a count and a fraction threshold, then the result of the SNP test is positive; otherwise, it is negative. The −1s indicating missing sequencing information do not contribute to the column totals used for computation of allele fractions. Note also that allele counts originating from the more accurate chromosomal and RNA sequence classes may be multiplied by a user-defined factor in order to increase their weights. Using this mechanism one may accept a sequence variation as real if it occurs either once in a single chromosomal or RNA sequence or at least twice in different ESTs.
The consecutive SNP tests applied to each column of the multiple alignment lead to a binary SNP status sequence. The SNPs in this sequence have to be mapped back to single bases because each SNP in the original alignment automatically generates k consecutive polymorphic positions in the k-tuple alignment. This is achieved by applying the same type of SNP test to the single-base alignment obtained after gap treatment. Only SNP positions that are positive in both tests are retained.
In the last postprocessing step, the SNP status sequence is scanned to identify clusters of SNPs separated by less than a user-defined number of bases. Such clusters, which may represent true multiple SNPs or errors in the input alignment, will be reported as single-candidate SNP regions in the output. The program offers two alternative output formats. The first one consists of the SNP status sequence in aFastAlign-like format, in which SNP candidate positions are represented by a user-defined character and all other positions by dashes. The second format comprises rich SNP reports for all candidate SNP regions detected including local multiple alignments as shown in Figure 1. This format can be easily hyperlinked to database sequence entries and chromatograms if corresponding identifiers can be extracted from the FastAlign header lines of the input files. The report format is intended for visual inspection by the SNP database curator. The algorithm implemented in snp_detect depends on a number of user-defined parameters, which are listed in Table4 along with their default values used in this work.
Command-Line Parameters of snp_detect Program
As a final step, the snp_detect output file was visually corrected to remove some alignments in which the same genomic sequence was present several times with different accession numbers, resulting in selected SNPs that did not fulfill our previously established criteria. We also eliminated some regions of low quality alignments that remained after the stringent matching parameters, in most cases a result of paralogous genes.
PowerBLAST
We performed a batch BLAST search of all known HC21 genes against all other human databases using PowerBLAST, a client-server program at NCBI. For this purpose we generated aFastAlign file containing 127 HC21 genes, which we used as input. More details on the program and the parameters used can be found at http://csnp.unige.ch/csnp_methods.html/.
Simulation of Amino Acid Substitutions
We estimated the proportion of conservative and nonconservative amino acid changes that would be produced by random nucleotide substitutions within 20 HC21 genes by simulation studies. For this purpose we extracted the cDNA sequences of the selected genes and introduced random mutations at a ratio of 2 : 1 transitions to transversions. We then translated the sequences using the expasy translate tool (http://www.expasy.ch/tools/dna.html) and generated amino acid alignments to identify changes. Amino acid substitutions were then classified as conservative or nonconservative using the PAM 250 matrix. Positive or neutral scores were considered conservative and negative scores nonconservative. A total of 200 repetitions were performed.
Verification of Data
To verify that candidate cSNPs were actually polymorphic, we designed primers around the putative cSNPs in order to generate short amplimers suitable for bidirectional sequencing. We PCR-amplified each region containing the candidate variant using pooled DNAs, as well as two controls.
Pools were made using DNAs from 10 unrelated individuals from CEPH families. The quantity of each DNA was measured by spectrophotometry and then normalized using SYBR Green (Applied Biosystems) real-time PCR amplification, to ensure that each DNA contributed equally to the pool. Sequencing was performed using an ABI 377XL automated sequencer (Applied Biosystems) and Big Dye terminator technology as recommended by the manufacturer.
Nucleotide Diversity Calculations
To calculate the nucleotide diversity parameter θ we used the formula θ = K/(L
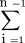 (1/i), where L is the length in base pairs, n is the number of chromosomes, and K is the number of variants found (Hartl and Clark 1997). For our calculations L was 379,187 bp, which represents the total length of HC21 genes as reported by Hattori et al. (2000). Because only ∼60% of the length of genes was actually covered by ESTs, however, the value of θ was corrected to take this
fact into consideration.
(1/i), where L is the length in base pairs, n is the number of chromosomes, and K is the number of variants found (Hartl and Clark 1997). For our calculations L was 379,187 bp, which represents the total length of HC21 genes as reported by Hattori et al. (2000). Because only ∼60% of the length of genes was actually covered by ESTs, however, the value of θ was corrected to take this
fact into consideration.
By looking at the multiple sequence alignments and the libraries of ESTs we estimated that for 95% of the genes n was between 6 and 40. We then took the median (18) for our calculations.
A flat file version of the database is available via anonymous ftp atftp://www.isrec.isb-sib.ch/sib-isrec/csnp/.
Acknowledgments
This work was supported by grants 31.57149.99 from the Swiss FNRS, 98-3039 from the OFES/EU, funds from the University and Cantonal Hospital of Geneva, and grants from the Ligue Genevoise Contre le Cancer, la Fondation Pour la Lutte Contre le Cancer, and la Fondation Dr. Henri Dubois-Ferrière Dinu Lipatti. We thank C. Rossier for assistance with cSNP confirmation and R.G. Lyle for bioinformatics advice and critical comments. We also thank Mick Drapper and members of the CERN AS-DH group at CERN for use of computers, and J. Michaud, M. Lalioti, K. Bucher, L. Bartoloni, M. Wattenhofer, M. Guipponi, J.L. Blouin, O. Menzel, and F. Chapot for contributing the gene-targeted cSNPs.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵6 Present address: Genetics and Bioinformatics Group, The Walter and Eliza Hall Institute of Medical Research, Post Office, Royal Melbourne Hospital, 3050 Victoria, Australia.
-
↵7 Corresponding author.
-
E-MAIL Stylianos.Antonarakis{at}medecine.unige.ch; FAX 41-22-702-5706.
-
Article published online before print: Genome Res.,10.1101/gr.164901.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.164901.
-
- Received September 13, 2000.
- Accepted November 21, 2000.
- Cold Spring Harbor Laboratory Press








