
Genomic Sequence Analysis of Fugu rubripes CFTR and Flanking Genes in a 60 kb Region Conserving Synteny with 800 kb of Human Chromosome 7
Abstract
To define control elements that regulate tissue-specific expression of the cystic fibrosis transmembrane regulator (CFTR), we have sequenced 60 kb of genomic DNA from the puffer fish Fugu rubripes (Fugu) that includes the CFTR gene. This region of the Fugu genome shows conservation of synteny with 800-kb sequence of the human genome encompassing the WNT2, CFTR, Z43555, and CBP90 genes. Additionally, the genomic structure of each gene is conserved. In a multiple sequence alignment of human, mouse, andFugu, the putative WNT2 promoter sequence is shown to contain highly conserved elements that may be transcription factor or other regulatory binding sites. We have found two putative ankyrin repeat-containing genes that flank the CFTR gene. Overall sequence analysis suggests conservation of intron/exon boundaries betweenFugu and human CFTR and revealed extensive homology between functional protein domains. However, the immediate 5′ regions of human and Fugu CFTR are highly divergent with few conserved sequences apart from those resembling diminished cAMP response elements (CRE) and CAAT box elements. Interestingly, the polymorphic polyT tract located upstream of exon 9 is present in human and Fugu but absent in mouse. Similarly, an intron 1 and intron 9 element common to human and Fugu is absent in mouse. The euryhaline killifish CFTR coding sequence is highly homologous to the Fugusequence, suggesting that upregulation of CFTR in that species in response to salinity may be regulated transcriptionally.
[The sequence data described in this paper have been submitted to the GenBank data library under accession no. AJ271361, for the combined cosmids 159C9, 146H13, 6M15, and 145M20.]
The puffer fish Fugu rubripes(Fugu) has one of the smallest genomes (400 Mb) of all vertebrates, attributable to a compactness of introns, intergenic distances, and marked reduction of repetitive DNA sequences. Because the Fugu genome possesses a gene repertoire similar to that of other vertebrates, it is a valuable model for vertebrate gene analysis (Brenner et al. 1993).
The utility of Fugu genome analysis for the identification of candidate genes at disease loci and the demonstration of conserved synteny between species is well established (Aparicio et al. 1995;Armes et al. 1997; Baxendale et al. 1995; Gellner et al. 1999; Sandford et al. 1997; Schofield et al. 1997; Venkatesh et al. 1997; Yeo et al. 1997). It is hypothesized that the large evolutionary distance separating Fugu and mammals (about 350 million years) will have resulted in divergence of most sequences except for those of conserved functional or structural importance, such as coding and regulatory regions. Previous comparative sequence analyses between the genomes of Fugu and other species have identified sequences important for the control of gene expression (Aparicio et al. 1995; How et al. 1996; Marshall et al. 1994; Sandford et al. 1997; Venkatesh et al. 1996; Venkatesh et al. 1997). Using this approach, Gellner and Rowitch (Gellner et al. 1999; Rowitch et al. 1998) identified potential regulatory elements for wnt1, which encodes a protein expressed in the developing midbrain.
However, levels of conservation in noncoding regions can vary considerably between Fugu and other species. Comparison of theFugu and human sequences in the Huntington's disease genomic region has not identified any conserved regulatory sequences (Baxendale et al. 1995). In contrast, in the region encompassing the WT1, Pax6 and RCN1 genes, there is significant noncoding homology betweenFugu and human at the PAX6 locus, implying conservation of regulatory elements (Miles et al. 1998). However, human WT1 generates 16 protein isoforms (Miles et al. 1998), whereas the sequence data predicts that Fugu WT1 will only produce two isoforms.
Synteny is not always conserved between Fugu and mammalian genomes. In the Surfeit gene cluster, there is extensive rearrangement of genes between Fugu and human within a region of otherwise conserved synteny. This suggests that intrachromosomal rearrangements, probably inversions, have occurred during evolution (Gilley et al. 1997; Gilley et al. 1999). Despite these caveats, there is ample evidence that Fugu sequence analysis provides important information about regulatory elements, conserved synteny, splicing, and gene organization (Angrist 1998; Coutelle et al. 1998; Gellner et al. 1999; Gilley et al. 1999; How et al. 1996; Maheshwar et al. 1996;Marshall et al. 1994; Miles et al. 1998; Trower et al. 1996; Venkatesh et al. 1996; Yeo et al. 1997).
Our aim is to produce gene therapy vectors for cystic fibrosis (CF) that generate tissue-specific expression of the cystic fibrosis transmembrane conductance regulator (CFTR) at physiologic levels. We therefore require a thorough understanding of CFTR gene structure and regulation. The CFTR gene is known to be expressed in a tightly regulated fashion (Chou et al. 1991; Denamur et al. 1994; Matthews et al. 1996; Pittman et al. 1995; Trapnell et al. 1991; Yoshimura et al. 1991a,b). The control and enhancer regions for CFTR are not yet fully defined, although DNase I hypersensitive sites (DHSs) have been found at − 20.5 kb, − 79.5 kb, 185 + 10 kb within the first intron (Smith et al. 1995; Smith et al. 1996), and 4574 + 15.6 kb beyond the 3′ end of the gene (Nuthall et al. 1999). Putative cAMP response elements (CREs), Y-box, Ap-1, Sp-1, major and minor transcriptional start sites, and CAAT-like sequences have been identified by sequence analysis, 5′ RACE, mutational analysis, and electrophoretic mobility shift assays (Chou et al. 1991; Denamur et al. 1994; Imler et al. 1996; Matthews et al. 1996; Pittman et al. 1995; Vuillaumier et al. 1997; White et al. 1998; Yoshimura et al. 1991a).
To identify conserved elements that regulate CFTR expression, we have isolated and cloned Fugu CFTR. Using comparative sequence analysis, we identified regions of conserved synteny and putative exon/intron boundaries for the CFTR and flanking genes.
RESULTS AND DISCUSSION
Isolation of Fugu CFTR Cosmids
In search of sequences that control tissue-specific expression of CFTR, we identified and cloned Fugu CFTR with sufficient flanking sequence to encompass 5′ and 3′ control elements and neighboring genes. We screened a Fugu cosmid library using degenerate oligomers, compiled from an alignment of killifish, human, dogfish, mouse, and Xenopus coding sequences, as hybridization probes. Two separate hybridization experiments were performed and only those cosmids that gave a positive signal to both experiments were investigated further. We observed a weak signal in a common set of nine cosmids in both hybridization experiments and found them to be related by restriction analysis, Southern transfer, and hybridization to human CFTR probes. Preliminary sequence of one cosmid was highly homologous to exon 13 of killifish CFTR (data not shown). These data confirmed the isolation of a set of cosmids covering part of the Fugu CFTR region.
Initially, we subcloned one cosmid 159C9 into a pBluescript vector and made four libraries containing either AluI or Sau3a cosmid insert DNA. Larger EcoRI and PstI inserts were also generated for subcloning and sequencing. Finally, we used cosmid walking to fill gaps in the sequence. The consed sequence assembly package was used to contig the sequence data (Ewing et al. 1998a,b). We sequenced cosmid 159C9 entirely, which was shown to contain candidate Fugu CFTR exons 3–24 with extensive 3′ sequence (∼29 kb). The insert ends of the other eight related cosmids were sequenced to find clones that would extend the sequence. Three cosmids, 146H13, 6M15, and 145M20, had additional sequence at the 5′ end and were used to complete theFugu CFTR genomic sequence by cosmid walking. Cosmid end sequencing (data not shown), restriction analysis, Southern transfer, and hybridization experiments gave valuable information on the wider genomic organization of the region, complementing the CFTR sequencing data.
Conservation of Synteny
In this study, conservation of synteny in the CFTR region has been demonstrated to extend over more than 800 kb of human and 60 kb ofFugu genomic sequence. In particular, we found that orthologs of genes flanking human CFTR, WNT2 (Monkley et al. 1996) (which belongs to a large gene family encoding a group of secreted signalling molecules), Z43555 (a protein of unknown function), and CBP90 (Ohoka et al. 1998) (a brain-specific cortactin-binding protein) are conserved in order and orientation in Fugu (Fig. 1). Using exon 1 of WNT2 and exon 2 of CBP90 as anchor points between species, this corresponds to a 9.2-fold genomic compaction inFugu relative to human.
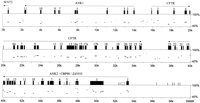
Genomic conservation and organization. Percentage identity plot (PIP) analysis of Fugu CFTR genomic region compared with the equivalent human region. Protein-coding exons are indicated by black rectangles. Gray and white boxes indicate 0.75 and 0.60 CpG:GpC ratios, respectively. Horizontal lines show blocks of homology between the Fugu and human sequences. Only blocks of homology occurring in the same relative order in both sequences are indicated by horizontal lines. Genes identified within theFugu genomic sequence are annotated and transcriptional orientation indicated above the appropriate exons. Exons of genes are numbered where space permits.
Multiple sequence alignment to 192 nt upstream of the putativeFugu translational start site for WNT2 shows 48% identity of residues between human, mouse, and Fugu. In comparison, exon 1 alignment gives 58% identity. Short regions of locally high homology may represent binding sites for transcription factors or other regulatory elements. The presumed translational start site resembles the Kozak (Kozak 1996) consensus, and is highly conserved between each of the three species (10 nt perfect conservation). The WNT2 promoter appears to be a “housekeeping” type of promoter with elements conserved through evolution.
Interestingly, in Fugu we identified regions with ankyrin repeat homology on either side of the CFTR gene (Fig. 1). The ankyrin repeat homology 5′ to CFTR is described in mouse (designated MMU_Orf3; Ellsworth et al. 2000). Of the 13 predicted (Genscan)Fugu exons, 11 shared conservation with human and mouse as revealed by Dotter analysis. Complete intron/exon structures for the genes were determined by Genewise (E. Birney, unpubl.). The entire open reading frame (392 aa) of a hypothetical protein (ANK1 inFugu) has been reconstructed. It contains four tandem ankyrin repeats and a Sterile Alpha Motif (SAM) domain (Pfam analysis), a similar domain arrangement to Tankyrase (a telomere-localized poly ADP-ribose polymerase; Smith et al. 1998).
There is a cluster 3′ to CFTR containing CBP90, ANK2, and Z43555 (Fig. 1). Genscan analysis of Fugu predicts six exons that share homology and correlate well with the exon/intron structure of the incomplete human coding region Z43555 (TREMBL accession no. 043388). A further two C-terminal exons, including an in-frame stop codon (Genscan), were predicted in Fugu and show conservation up to the stop codon in human sequence. ANK2 lies between CBP90 and Z43555 (Fig. 1) and contains 12 predicted exons (NIX;http://www.hgmp.mrc.ac.uk/NIX/) encoding putative ankyrin repeat motifs (Pfam analysis). The close proximity and consistency of orientation inFugu and human, as well as an absence of potential polyadenylation signals (AAUAAA) within the region in Fugu, suggest that CBP90, ANK2, and Z43555 may all represent fragments of the same gene. It remains possible that one or more of the predicted exons are spurious. Further cDNA analysis is required to confirm the genetic structure. For supplementary information, seehttp://www.hgu.mrc.ac.uk/users/Heather.Davidson/Fugu.html. The comparison of relatively conserved coding sequences superimposed on diverged sequence background in Fugu, human, and mouse demonstrates the utility of Fugu: mammal comparison for the identification of putative novel genes.
Fugu Versus Mammalian CFTR
At the predicted amino acid level, a global alignment ofFugu and human CFTR demonstrates 58% identity and 75% similarity. In comparing human and Fugu functional CFTR domains, NBD1 (nucleotide-binding domain) exhibits 71% identity and 87% similarity, NBD2 58% identity and 73% similarity, the R (regulatory) domain 46% identity and 63% similarity, and MSD1 (membrane-spanning domain) 61% identity and 76% similarity, and MSD2 59% identity and 73% similarity. The fourth extracellular loop encoded within exon 15 and three regions of the R domain display a relatively high level of sequence divergence (Fig.2).

Sequence homology and conservation of exon/intron boundaries. Alignment of human, mouse, killifish, and Fugu CFTR. The triangles mark approximate intron/exon boundaries. Domains of CFTR are as indicated: NBD = nucleotide-binding domain; R-domain = regulatory domain; TM = transmembrane segment. Horizontal lines linking transmembrane segments represent extracellular loops. Diamonds above the phosphorylated residues predict dibasic cAMP-dependent phosphorylation sites. Short horizontal bars marked with asterisks above exon 15 indicate glycosylation sites. In the background shading at each position, phosphorylation and glycosylation sites: black indicates perfect homology, dark grey three out of four, and light grey two out of four residues matching.
Within the R domain of human CFTR, there are nine dibasic consensus phosphorylation sites for cAMP-dependent phosphorylation, a process critical for the regulation of CFTR Cl channel activity (Riordan et al. 1989; Xiu-Bao et al. 1993). All but two of these sites are conserved inFugu CFTR (Fig. 2). Of the remaining sites, serine 700 is converted to a monobasic consensus phosphorylation site, while threonine 788 is abolished in Fugu CFTR. Interestingly, serine 670, a monobasic consensus phosphorylation site in human CFTR, is dibasic in Fugu, killifish, and mouse. Of the two N-glycosylation sites within the fourth extracellular loop of human CFTR, only the C-terminal site is predicted (Bause 1983) to be glycosylated in Fugu and killifish (Fig. 2).
The alignment of human, mouse, and Fugu CFTR genomic sequences as guided by the predicted amino acid sequences suggests that the relative position and coding phase of exons is conserved. Such conservation of exon position and phase suggests that equivalents of all known human splice forms could be generated from the Fuguortholog. For each exon, the 40-nt flanking splice donor and acceptor sites from both Fugu and human CFTR were isolated and compared directly. Although conservation was calculated for both intron and exon components of each alignment, no correlated divergence from core splice consensus sequences was found. Interestingly, the Fugu intron 9 splice acceptor region contains 5′-TTTTTTTT-3′, possibly equivalent to the polymorphic polyT tract that affects the variable in-frame skipping of exon 9 of human CFTR (Chu et al. 1991, 1993). However, this polyT tract is absent in mouse (Ellsworth et al. 2000).
The 3′ splice site at the intron 9/exon 10 junction of CFTR is a good match with the consensus YYYnNYAG/− (Moore 2000) and is conserved between human, mouse, and Fugu (Fig.3A). Within the intron and nearly equidistant (17–18 nt), in both human and Fugu, from the splice junction, there is a block of conservation (13–14 nt) whose position and resemblance to the consensus makes it a good candidate to be the splice branch site. The equivalent mouse sequence also contains consensus splice branch point homology, but the block of homology shared between human and Fugu is specifically absent in the mouse (Fig. 3A). The level of conservation and consistency of position makes this an intriguing observation, though its significance is unclear.

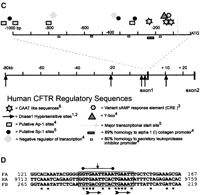
Putative human CFTR regulatory domains. Relative positions of proposed human CFTR transcription regulatory sequences and their conservation between mammals. (A) CFTR Intron 9/exon 10 3′ splice site aligned in mouse, human, and Fugu. Bold type indicates blocks of sequence conserved between human andFugu, but not in mouse. Italic sequence shows consensus branch-point and 3′ splice site sequences. The proposed branch-point A is marked with *. $ indicates the first nucleotide of exon 10. Capitalized letters of consensus sequences summarize positions where both Fugu and human match the consensus. (B) Alignments 1 to 4 illustrate the conservation of selected CFTR promoter elements between human (Homo sapiens), gibbon (Hylobates lar), monkey (Saimiri sciureus), cow (Bos taurus) and rabbit (Oryctolagus cuniculus) at coordinates relative to the start of translation of human CFTR. Asterisks indicated nucleotides conserved between all aligned species. Background shading marks the extent of proposed elements. (C) The relative position of sequence motifs implicated in CFTR regulation, DNase I hypersensitive sites, and exons one and two are indicated with respect to the translational start site (ATG). Annotated elements are as defined in: 1(Smith et al. 1995), 2(Smith et al. 1996), 3(Matthews et al. 1996), 4(Chou et al. 1991), 5(Yoshimura et al. 1991a), 6(Pittman et al. 1995). (D) FA and FBFugu CFTR intron 1 elements are shown in alignment with the conserved human HA element. Asterisks indicate nucleotides conserved in all three elements. The 10-bp perfect palindromes of FB and HA are indicated by a horizontal bar, and the axis of symmetry is indicated by a vertical arrow. Horizontal arrows indicate partially conserved direct repeats in each element. The coordinates indicate distance from the 3′ end of CFTR exon 1. Background shading marks the extent of the proposed element.
CFTR Transcriptional Regulation
Some transcription control elements of the CFTR gene are currently defined solely by homology to short consensus sequences. Our interest is to identify these elements which are functional and incorporate them into genomic context vectors for CF gene therapy. Little is currently known about the general sequence conservation and particular features of housekeeping gene promoters conserved between distantly related vertebrates.
Multiple sequence alignment (ClustalW; Thompson et al. 1994) of sequences directly upstream of CFTR coding sequence (data not shown) demonstrates good sequence conservation between mammals (Vuillaumier et al. 1997), but not between mammals and fish. Within the CFTR core promoter region of the aligned mammalian species, previously proposed regulatory sequence elements are generally poorly conserved (Fig. 3B). The Sp-1 sites, Ap-1 sites, and putative negative regulators of human CFTR (Fig. 3C; Chou et al. 1991; Denamur et al. 1994) are not highly conserved between salmon, Fugu, killifish, primate, bovine, or rabbit, questioning their importance in core promoter activity and tissue-specific expression (Fig. 3B). We have found no Sp-1 or Ap-1 sites within the Fugu CFTR promoter region, as has been described for the Fugu Surfeit family of genes (Gilley et al. 1997, 1999), which have housekeeping promoters similar to CFTR. Moreover, a TATA box at –545 bp in the CFTR promoter region is unique to Fugu CFTR. These sequence alterations may represent genuine differences in housekeeping gene regulation between mammals and fish.
Blocks of conserved promoter sequence in CFTR from mammals suggest functional significance, but the equivalent Fugu regions are again devoid of recognizable homology to these sequences (data not shown). However, homology to a CRE (TGACGTCA; Matthews et al. 1996) is found in Fugu at position –282 bp (TGACGT), while slight homology to an inverted Y box (consensus CWGATTGGYCYA) is found at position –344 bp (CAGATTCTATAT). The inverted and imperfect CAAT box (AATTGGAAGCAAAT) with conserved residues TTGGAAGCART (found in human, two primates, cow, and rabbit) is not completely conserved inFugu, although at position –174 bp, there is the well-conserved GAGGAGAAGCAAGA motif. In mammalian species, the CRE and Y box normally overlap, whereas in Fugu, they do not. These data highlight the highly divergent nature of the Fugugenome and the lack of conserved regulatory elements between human and Fugu in the CFTR promoter region.
In the wider genomic context, no substantial blocks of conserved sequence (phylogenetic footprints) were identified in proximity to CFTR. We found no substantial regions of homology when we performed percentage identity plot (PIP) pairwise comparisons betweenFugu and mouse and Fugu and human genomic sequences (Fig. 1; http://www.hgu.mrc.ac.uk/users/Heather.Davidson/Fugu.html). PIP analysis, however, supports the human exon predictions for the two ankyrin repeat-containing proteins, Z43555 and CBP90. In addition to the predicted exons of CFTR and flanking genes, PIP analysis suggested several other regions of potential conservation (unannotated horizontal lines in Fig. 1); of these, all in the CFTR region can be attributable to regions of low compositional complexity, producing multiple spurious hits when the PIP chaining and single coverage models are not used (see Methods). Of particular interest is the absence of detectable homology in regions previously identified as containing DHSs (Nuthall et al. 1999; Smith et al. 1995, 1996). The DHSs at −79.5 and −20 kb show weak correlation with CFTR expression in both cultured epithelial cell lines and primary duct epithelial cells, and we found no evidence for their conservation in Fugu (Smith et al. 1995). The DHS at –79.5 in human will place it in the middle of the ankyrin repeat-containing protein, which has yet to be proven to be functional, and therefore this DHS site may not be involved in CFTR regulation.
The DHS in intron 1 (position 181 + 10 kb; Smith et al. 1996) correlates well with the levels of CFTR expression in these cell lines (Smith et al. 1995). Inclusion of human CFTR intron 1 has also been directly shown to confer transcriptional upregulation in a controlled reporter system (Mogayzel and Ashlock, 2000). Interestingly, we have found a complex element in Fugu intron 1 which shows noncoding homology between the human and Fugu species. This intron 1 element is too short to be picked up by PIP, but was found using dot matrix methods (data not shown;http://www.hgu.mrc.ac.uk/users/Heather.Davidson/Fugu.html). The element contains palindromic and direct repeat features (Fig. 3D). However, in the orthologous mouse genomic sequence (Ellsworth et al. 2000), there is an insertion in this element which casts doubt on its significance. It overlaps a known cluster of DNase I hypersensitive sites and DNase I footprints in human CFTR that correlate well with CFTR expression. Specifically, we have found a 17-bp element conserved between human intron 1 element A (HA) and Fugu intron 1 element A (FA). FA is conserved at 13/17 nucleotides between human andFugu (Fig. 3D), corresponding to coordinates 181 + 9.727 kb and 181 + 135 bp, respectively. The orientation of FA is conserved with respect to CFTR transcription. Interestingly, within the Fugu intron 1, there is a second, inverted copy (Fugu intron 1 element B [FB]) 80 bp downstream. FB in Fugu shares 11/17 nucleotides with FA and 14/17 nucleotides with the human HA (Fig. 3D). However, no human equivalent of FB was identified.
At the 3′ end of the human CFTR gene, there is a DHS at position 4574 + 15.6 kb that regulates tissue-specific expression of CFTR (Nuthall et al. 1999). There is no evidence of conservation of this site in Fugu. This DHS and/or the other DHSs (at 4574 + 5.4 − 7.4 kb; Nuthall et al. 1999), which do not regulate CFTR expression, might instead regulate the expression of the Z43555 and CBP90 (two genes located 48 kb and 160 kb 3′ from the end of the CFTR gene).
Killifish CFTR Regulation
The euryhaline killifish (Singer et al. 1998) adapts rapidly to extreme changes in salinity. Exposure of freshwater-adapted killifish to seawater increases expression of killifish CFTR, implying a role for CFTR in salinity adaptation. Despite Fugu not being similarly adapted, its CFTR protein sequence is highly homologous (84% identity and 91% similarity) to that of killifish. Therefore, the CFTR-mediated freshwater adaptation of killifish is likely to be solely explained by transcriptional upregulation rather than fundamental differences in the property of the channel.
Summary
Our study has shown conservation of synteny and orientation betweenFugu and human over a large, multigenic region. All genes identified appear to be capable of functioning in both species. Like CFTR, WNT2 has a housekeeping promoter. However, the WNT2 promoter has conserved elements between human, mouse, and Fugu, whereas there is little conservation in the promoter of Fugu CFTR. This suggests that regulation of WNT2 is far more conserved that that of CFTR. The data are consistent with complete conservation of CFTR exon/intron boundaries between Fugu and human, and there is extensive homology between functional domains. In noncoding regions of the CFTR gene, human and Fugu are highly divergent, and apart from a diminished CRE and CAAT box, the putative promoter regions are devoid of conserved regulatory domains. The inclusion of the mouse orthologous genomic sequence in the intron 1 comparison casts doubt on the validity of the identified conserved element identified in human and Fugu even though it is near a previously described DHS (Nuthall et al. 1999). The element's importance must be determined empirically to evaluate its functional significance. However, the additional finding of the polyT tract and the intron 9 element, both of which are present in human and Fugu but absent in mouse, suggests that CFTR regulation in the mouse lineage has evolved eccentrically. There may be a correlation between these observations and the known differences between mouse and human in CFTR expression patterns (Manson et al. 1997) and mutant phenotypes (Davidson et al. 1995).
This and other studies (Ellsworth et al. 2000) of CFTR genomic structure and its conservation between species will inform our overall strategy of producing minimal genomic context vectors that provide regulated tissue-specific expression for CF gene therapy.
METHODS
Southern Blots and Hybridizations
The Fugu genomic cosmid library (coverage eightfold) was obtained from the Medical Research Council Human Genome Mapping Project Resource Centre. To test the degenerate oligomers, hybridization experiments at various temperatures and washing conditions were performed on a human CFTR P1 artificial chromosome (Ioannou et al. 1994), whose sequence was expected to be as divergent from the degenerate oligomer as that of a Fugu CFTR sequence. Conditions for the 53-mer degenerate oligomer (hybridization experiment 1) were optimized to be: hybridization at 60°C with three washes in 4× SSC (1× saline sodium citrate [SSC] is 0.15 mNaCl, 0.015 m sodium citrate), 0.1% sodium dodecyl sulphate (SDS), one at room temperature followed by two at 60°C. For the mixed oligomer hybridization (hybridization experiment 2), the conditions were optimized to be: hybridization at 48°C followed by three washes in 4× SSC, 0.1% SDS at room temperature, and one at 37°C. The Southern transfer blots of the restriction digestedFugu cosmid DNA were hybridized at 58°C and washed at 68°C three times in 2× SSC, 0.1% SDS.
The oligomers used in the hybridization were: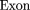
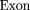
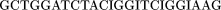
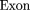
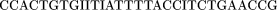
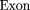
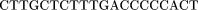
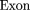
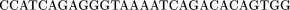
Cloning and Sequencing
The library was cloned into pBluescript vector, and subclones were sequenced with KS and SK primers (Stratagene Inc.) and ABI dye terminator chemistry using an ABI377 automated DNA sequencer. Sequences were assembled with the consed sequence assembly program (http://www.genome.washington.edu; Ewing et al. 1998a,b). Sequencing of cosmids with custom primers was used to close gaps and complete both strands. The sequence coverage for the Fugu CFTR genomic region is at least two high-quality sequence runs in both directions, coding regions being more extensively sequenced. In two noncoding regions, both situated between exon 18 and 19 of CFTR, short GC-rich tracts caused high-quality sequencing to be obtained in one direction only, despite several attempts. Another region we sequenced in one direction only is located at 21458 bp 3′ to the end of the coding sequence of CFTR between Z43555 and CBP90 (Fig. 3).
Comparative Sequence Analysis
Nucleotide sequences (Washington University Genome Sequencing Center) for human-derived bacterial artificial chromosomes were used to assemble 864 kb (from 7q31–32 ) of contiguous human genomic sequence, including and flanking the CFTR transcriptional unit (GenBank database accession nos. AC000061, AC000111, AC002465, AC003045, AC004240, andAC002431). Assembly was performed by iterative Fasta (Pearson et al. 1988) searches to determine overlapping coordinates, and final assembly was achieved using in-house software.
For human genomic sequence, a sliding window method of sequence fragmentation was used to generate an artificial contig of sequential 100-kb fragments with 50-kb overlaps. Each human fragment and theFugu sequence was analyzed using programs for feature prediction and homology searching of appropriate public databases, coordinated through the NIX interface (http://www.hgmp.mrc.ac.uk/NIX/). Putative exons and genes were compared at the nucleotide and encoded amino acid level to known genes and the dbEST database using theBLAST (Altschul et al. 1997), Fasta (Pearson and Lipman, 1988), and HMMer (Eddy 1996) homology search tools. Exact coordinates of coding sequence and intron/exon boundaries were predicted using the Wise2 package (Birney et al. 1996). Homology templates used in Wise2 splice site prediction were killifish CFTR, rat CBP90, human WNT2, and Z43555 amino acid sequences (TREMBL accession nos. O73677, O88864,P09544, and O43388, respectively).
We performed PIP (PipMaker http://globin.cse.psu.edu/pipmaker/) pairwise comparison between Fugu and human and Fuguand mouse genomic sequences. For PIP analysis, low-complexity regions of the Fugu sequence were masked using RepeatMasker (A. Smit and P. Green, unpubl.). PipMaker default settings were adjusted so that match = 1, transition = −0.6, transversion = −0.8, and alignment cutoff = 18. The comparisons were only made in the forward strand. Chaining and single-coverage models of alignment distribution were used to reduce spurious matches (http://globin.cse.psu.edu/pipmaker/pip-instr2.html).
The multiple nucleotide sequence alignment of mammalian CFTR promoters (data not shown) was performed using ClustalW (Thompson et al. 1994) with default parameters for nucleic acid alignment. Nucleotide sequences directly upstream of CFTR coding sequence were too divergent between fish and mammals to construct an informative multiple sequence alignment (data not shown). The alignment of human, mouse, killifish, and Fugu CFTR amino acid sequences (Fig. 2) was achieved using ClustalW (Thompson et al. 1994). Pairwise comparisons between human andFugu CFTR at the nucleotide and amino acid levels were carried out using ALIGN (Pearson and Lipman, 1988) default parameters. The GenBank accession codes for the various CFTR protein or DNA sequences were: human protein P13569, mouse protein P26361, killifish protein AAC41271, human promoter AC000111, mouse promoter L04873, cow promoterX95926, rat promoter X95927, squirrel monkey promoter X95928, salmon promoter AF155237, killifish promoter AF000271, crab-eating macaque gene X95929, gibbon gene X95930, rabbit gene X95931, andXenopus promoter X65256.
Fugu and human genomic sequences were directly compared after fragmentation with a window size of 10 kb and 1 kb using Fasta and Lalign (Pearson and Lipman 1988) with known coding sequence masked.Fugu genomic sequence was further fragmented into sequential 100-bp blocks with 50-bp overlap and compared to human sequences using Fasta. Further analyses used a pairwise dot-matrix analysis performed with the Dotter program (Sonnhammer and Durbin 1995) at window sizes of 25 and 45 nucleotides.
Acknowledgments
We thank Prof. Nicholas Hastie and Dr. David Sheppard for helpful comments on the manuscript, Stewart McKay and Agnes Gallagher for DNA sequencing, Webb Miller for help and advice with the optimization of the PIP analysis, and the United Kingdom Human Genome Mapping Project Resource Centre for supplying the Fugu cosmids. This work was supported by the UK Medical Research Council.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL H.Davidson{at}ed.ac.uk; FAX 44 131 651 1059.
-
- Received April 6, 2000.
- Accepted June 2, 2000.
- Cold Spring Harbor Laboratory Press








