
WABA Success: A Tool for Sequence Comparison between Large Genomes
Whole-genome sequence comparisons between bacterial sequences are one thing, but try comparing two eukaryotic genomes, each containing tens or hundreds of millions of nucleotides. And try to do it on your desktop machine in your office or at home. That is what Kent and Zahler (2000) have tried, and the results are presented in this issue of Genome Research. The use of evolutionary conservation to unveil functional information contained within genomes is not new. In the case of the nematode, comparisons of Caenorhabditis elegans to its close relative Caenorhabditis briggsae go back as far as Emmons et al. (1979). Snutch (1984) made the firstC. briggsae genomic library available to the research community. These two nematodes are almost identical in morphology and development, yet their genomes have been separated for a sufficient amount of time to allow intronic and intragenic sequences to become effectively randomized, while protein-encoding sequences andcis-linked regulatory elements are conserved. C. briggsae has diverged from C. elegans in regions of unselected sequence, the middle of large introns, between genes, and at the third nucleotide of synonymous codons in genes not abundantly translated. Based on the now-outdated concept of a constant evolutionary clock, these two species were estimated to have diverged some 30–60 million years ago. These facts prompted a C. briggsae genome sequencing effort to be initiated by The Washington University Genome Sequencing Center in St. Louis, Missouri. The project was given a jump-start with the construction of a physical map (Marra et al. 1997) and an open invitation to the research community to participate in the selection of fosmids for sequence analysis. Researchers from around the world probed microarrayed fosmid filters with their favorite gene and contacted The Genome Sequencing Center with a request to sequence the identified fosmid. Currently, approximately 10% of the genome has now been completed and is available athttp://www.genome.wustl.edu/pub/gsc1/sequence/st.louis/briggsae/finish/. Comparison of C. elegans and C. briggsaesequence has facilitated the identification of cis-linked regulatory elements (Heine and Blumenthal 1986), the cloning of genes as a result of their syntenic relationship (Kuwabara and Shah 1994), and interpretation of complex gene structure (Thacker et al. 1999). In many cases examined, adjacent genes are conserved both in position and orientation, with the occasional interruption caused by a transposable element or an apparent pseudogene.
Prior to the availability of C. briggsae sequence, gene feature identification was done by abinitio prediction. Many computer programs exist that attempt to predict the exon-intron structure of genes from genomic sequence. These programs vary in their accuracy and are at their worst when asked to predict the first and last exons of genes, often failing to identify correctly exons and introns outside the actual coding element. Messenger RNA and EST-based feature detection is often confounded by large transcripts, genes that have low levels of transcription, or genes lacking poly(A)+-containing transcripts. Further complications adding to the problem of gene finding include the still-murky rules of sequence identification unknown for numerous important genomic elements (snRNAs, ribozymes, transcription factor binding sites, replication origins, chromatin folding and packaging signals, meiotic pairing information, etc.). Because of these issues, the initial annotation of the C. elegans genome was done interactively using GENEFINDER, a program written by Phil Green and LaDeana Hillier (unpubl.). The program, which was ahead of its time, did a remarkable job of gene prediction. Taken together with manual interpretation, the cDNA data (Kohara 1996) and related genomic sequence data from C. briggsae, gene structure predictions were made for > 19,000 C. elegans genes.
Kent and Zahler (2000) have taken advantage of the availability of genomic sequence from these two closely related nematodes to test the feasibility of doing large-scale alignments between genomic DNA of different species. They have developed an algorithm for sequence comparison in which every third base (the wobble position of the codon) is ignored. The wobble-aware bulk aligner (WABA) allows the sensitive identification of conserved coding regions. They use this algorithm in a three-tiered process to identify conserved regions between eight million base pairs of C. briggsae genomic sequence and the entire 97 million base pairs of C. elegans. Their analysis was performed on a readily available 450 mHz Intel-based machine. The results they have achieved are remarkable and will provide a useful resource for all in the continuing analysis of genomes. One of the examples given by the authors is the bli-4 (K04F10.4) gene. The structure of this gene was characterized previously by examination of the conservation between C. elegans and C. briggsae (Thacker et al. 1999; Fig. 1B). Traditionally, gene prediction programs have been forced to ignore the computationally complex problem of ab-initio prediction of splice variants. Neither ACEDB nor the Genie program predict the entire nine alternate transcripts described in Thacker et al. (1999). Figure 1A shows the WABA/intronator display for the gene. The areas of C. briggsaehomology identified by the WABA program would greatly assist researchers by alerting them to the strong possibility that alternative transcripts may exist. The intronator display brings together gene predictions, C. briggsae homologies, and cDNA information in an easily used and intuitive fashion.

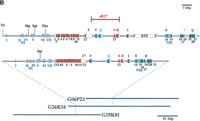
(A) Exon prediction in genomic regions near bli-4(also called K04F10; from Kent and Zahler athttp://www.cse.ucsc.edu/∼kent/cgi-bin/tracks.exe?where=K04F10). (B) Schematic representation of the bli-4 gene fromC. elegans (top) and C. briggsae(bottom). The common region(s) encoding the signal peptide, prodomain, protease domain, and middle domain are shown in brown. Alternatively spliced exons that encode carboxyl termini unique to the individual isoforms are labeled alphabetically and color coded. The position and extent of the e937 3325-bp deletion is indicated. Open boxes represent noncoding exons or untranslated regions; shaded boxes represent coding exons. Hatched boxes (labeled I–VIII) represent regions of nucleotide homology that may constitute regulatory elements, particularly those at the 5′ end. (Xb) XbaI; (Blp) BlpI; (Bgl) BglII; (Xho) XhoI. The approximate location of the C. briggsae bli-4 gene is indicated by dashed lines on the fosmid clones used in Thacker et al. (1999). The entire sequence of clones G25K01 and G06P23 has been determined, whereas G26K16 was used solely for transformation rescue experiments. (Reprinted with permission from Thacker et al. 1999.)
One surprising observation made by Kent and Zahler (2000) was the shortness of the syntenic segments, which is partly a consequence of the length of the C. briggsae clones. The number of fragments that clones are broken into by the alignment exhibited a bimodal distribution, which corresponds to some extent to the position of the sequence on the C. elegans autosomes. In the gene-rich central clusters, long alignments were observed, predicted to constitute approximately 40% of the genome. In contrast, the flanking arms were more susceptible to rearrangement. It has been known for some time that meiotic recombination is much higher on the chromosome arms. This fact and the observation that sequence similarities to organisms other than nematodes tends to be lower on the arms led the C. elegansSequencing Consortium (1998) to suggest that the DNA in the arms might be evolving more rapidly than that in the central regions of the autosomes.
Computational tools, like WABA, will be essential for future analysis of large vertebrate genomes. Computational comparative genomics is expected to become an integral part of the analysis of human and mouse genomes and will be needed to identify coding elements and other functional components resistant to analysis by more conventional genetic and molecular approaches.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL arose{at}gene.nce.ubc.ca; FAX (604) 822-5348.
- Cold Spring Harbor Laboratory Press








