
Rice Transposable Elements: A Survey of 73,000 Sequence-Tagged-Connectors
Abstract
As part of an international effort to sequence the rice genome, the Clemson University Genomics Institute is developing a sequence-tagged-connector (STC) framework. This framework includes the generation of deep-coverage BAC libraries from O. sativa ssp.japonica c.v. Nipponbare and the sequencing of both ends of the genomic DNA insert of the BAC clones. Here, we report a survey of the transposable elements (TE) in >73,000 STCs. A total of 6848 STCs were found homologous to regions of known TE sequences (E<10−5) by FASTX search of STCs against a set of 1358 TE protein sequences obtained from GenBank. Of these TE-containing STCs (TE–STCs), 88% (6027) are related to retroelements and the remaining are transposase homologs. Nearly all DNA transposons known previously in plants were present in the STCs, including maize Ac/Ds,En/Spm, Mutator, and mariner-like elements. In addition, 2746 STCs were found to contain regions homologous to known miniature inverted-repeat transposable elements (MITEs). The distribution of these MITEs in regions near genes was confirmed by EST comparisons to MITE-containing STCs, and our results showed that the association of MITEs with known EST transcripts varies by MITE type. Unlike the biased distribution of retroelements in maize, we found no evidence for the presence of gene islands when we correlated TE–STCs with a physical map of the CUGI BAC library. These analyses of TEs in nearly 50 Mb of rice genomic DNA provide an interesting and informative preview of the rice genome.
Transposable elements (TEs) are ubiquitous in all organisms (Burge and Howe 1989; Xiong and Eickbush 1990). In plants, TEs are classified into two main classes (Flavell et al. 1994). Retrotransposons comprise Class I and transpose via an RNA intermediate. Class I TEs include retrotransposons with long terminal repeats (LTRs) such as Ty1/Copia-like and Ty3/Gypsy-like, as well as non-LTR retrotransposons. The class II TEs transpose via a DNA intermediate and in plants have been found mainly in maize. Class II TEs include Ac/Ds,En/Spm, and Mutator (Federoff 1989). MITEs, that is, miniature inverted-repeat transposable elements, such as maizeTourist and Stowaway, fall into a newly described third class of TEs (Bureau and Wessler 1992, 1994a,b, 1996). The mechanism of transposition of MITEs is still unclear, although they have received considerable attention recently due to their high copy numbers and tendency to be associated with genes in maize (Wessler et al. 1995; Zhang et al. 2000).
Rice (Oryza sativa) is the main staple food for more than half of the world's population and is of great economic importance. Among the cereal grasses, rice has the smallest genome size (430 Mb) and, as revealed by comparative mapping, has substantial conservation of synteny with other cereal crops such as maize, sorghum, and wheat (Gale and Devos 1998). Consequently, rice is an ideal representative for cereal genomics studies and is the focus of an international effort to completely sequence its genome. Although numerous TEs have been reported in rice, no comprehensive investigation has been carried out on a genome-wide scale, because the majority of rice TEs were uncovered by chance or by limited assays using conserved regions such as reverse transcriptase of retrotransposons (Hirochika et al. 1992; Motohashi et al. 1996; Kumekawa et al. 1999). As part of the International Rice Genome Sequencing Project (IRGSP), a rice BAC library was constructed from a partial HindIII digest of the genome of the rice variety Nipponbare (Budiman 1999), and the ends of BAC clone inserts have been sequenced. BAC end sequences will serve as sequence-tagged-connectors (STCs) for selecting minimum overlapping clones for genome sequencing (Venter et al. 1996).
The generation of >73,000 Nipponbare STCs also provides an opportunity to preview TE content and distribution in rice genome. The current STC library contains ∼48 Mb of rice genomic DNA after vector removal, with an average sequence read of 707 nucleotides. With an average insert of 128.5 kb, the CUGI rice BAC library is expected to cover ∼10 rice genome equivalents. Preliminary efforts to confirm the coverage of the library based strictly on sequence comparison of the STCs to finished rice BACs have shown that the estimated coverage is ∼10.4 genome equivalents (data not shown). Assuming that theHindIII sites are evenly distributed, our 73,000 STCs should be distributed one STC every 9 kb across the 430-Mb rice genome.
TEs are one of the major sources of repetitive sequences in cereal plants and have been a concern of the IRGSP as a potential source of problems in completing the rice genome sequence. Here, we report the TE content of the STC database and show that the rice genome probably contains a small fraction of TEs in comparison with other cereal genomes, such as maize. The small amount of TEs confirms rice as a well-chosen model crop genome. We note the discovery of several potentially novel TEs, and we investigate the location of TE–STCs on the current physical map of the CUGI rice BAC library. We find that the TEs appear to be randomly distributed with respect to potential genes, identified by similarity to rice ESTs.
RESULTS
TE Content of STC Library
To analyze the number and types of TE-like elements present in the STC database, we used FASTX (Pearson et al. 1997) to compare 73,362 BAC end sequences (STCs) with a set of 1358 TE protein sequences. At an expectation cut-off value of 10−5 or less, 6848 STCs were found to contain regions of homology to known transposable elements. The vast majority of STCs (6027) are homologous to retrotransposons, whereas the remaining 821 are homologous to various transposases of class II transposons (Table 1). STCs homologous to retrotransposons were further classified as Gypsy-like (4124),Copia-like (1401), and non-LTR (502) on the basis of classification of the most similar protein sequences. To assess the accuracy of our retrotransposon classification, we used TFASTX to search the STC database with protein sequences of representativeGypsy (rice RIRE2), Copia (maize Hopscotch), and non-LTR (rice CAA73800) retrotransposons as query sequences. For all three searches, we found a total of 1959 STCs with significant similarity (E<10−5). Divided by retrotransposon classification, the proportions of STCs identified in each class for both the FASTX and TFASTX searches were nearly identical (Fig.1).
Transposable Element Content of the Rice STC Database
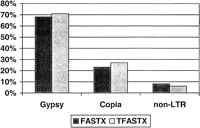
Classification of retrotransposons identified by FASTX and TFASTX searches. Fractions shown are percentages of total retrotransposon-containing STCs. FASTX searches were conducted using the rice STC database as queries to search the 1358-member TE database. Classification as gypsy, copia, or non-LTR was made on the basis of the most similar transposable element protein sequence. TFASTX searches were conducted using Gypsy-like rice RIRE2 (BAA84458, 1397 homologous STCs), Copia-likeHopscotch from maize (T02087, 528 homologous STCs), and a rice non-LTR LINE (CAA73800, 119 homologous STCs) as queries to search the STC database.
As a control, we performed an identical survey on 16,360Arabidopsis STCs sequenced by Genoscope (http://www.genoscope.cns.fr/externe/arabidopsis/data/bac_ends) and compared the results from both species with the publicly available chromosomal sequences. In our FASTX survey of the ArabidopsisSTCs, we found 1197 and 143 STCs homologous to retroelements and transposases, respectively. Although the actual numbers differ, the proportions of TEs in the rice and Arabidopsis STC databases are nearly the same, with 8.2% of the Arabidopsis STCs and 9.3% of the rice STCs showing homology to a TE. Within each species, retroelements account for 89.3% of Arabidopsis TE–STCs and 88.0% of rice TE–STCs (Fig. 2). The TE content of the chromosomal sequences from each plant shows slightly different proportions. The annotation of Arabidopsis chromosome 2 identified 563 TEs with 404 (71.7%) retroelements (Lin et al. 1999). Similarly, a survey of a 1-Mb PAC contig from rice chromosome 1 sequenced by the Rice Genome Research Program (http://www.dna.affrc.go.jp:82/genomicdata/GenomeFinished.html) revealed 68 unique regions homologous to TEs in TFASTX searches with the proteins of our 1358-member TE database. Of these 68 unique TE-like regions, 66.1% are homologous to retroelements (Fig. 2).
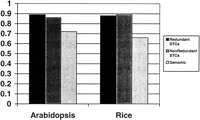
Proportions of retroelements found in redundant STCs, nonredundant STCs, and genomic sequences from Arabidopsis and rice. Transposable element homologies were identified as described in text. Classification of Arabidopsis chromosome 2 transposable elements was obtained from the chromosomal annotation (Lin et al. 1999). Total observed homologs are as follows: Nonredundant STCs: 350 rice transposases, 2754 rice retroelements; 101 Arabidopsistransposases, 628 Arabidopsis retroelements. Redundant STCs: 821 rice transposases, 6027 rice retroelements; 143Arabidopsis transposases, 1197 Arabidopsisretroelements. Genomic DNA: 23 rice transposases, 45 rice retroelements; 159 Arabidopsis transposases, 404Arabidopsis retroelements.
On the basis of these results, it is clear that the proportions of retroelements present in both the Arabidopsis and rice STC databases are slightly higher than preliminary estimates of the actual genomic content. The over-representation of retroelements is not likely to be the result of errors in the FASTX analysis, as the TEs of the 1-Mb rice PAC contig was analyzed in a similar way (TFASTX) and also showed a lower proportion of retroelements than identified in the rice STCs. Further, if we eliminate STC redundancy by examining only STCs that are <95% identical to each other, we find 729 TE–STCs inArabidopsis (628 of which are retroelements) and 3104 TE–STCs in rice (2754 of which are retroelements). In both the redundant and nonredundant STC analyses, the ratio of retroelements to transposases is ∼9 to 1 (Fig. 2). Thus, the over-representation of retroelements appears to be inherent to both STC databases and may be due to cloning-site bias.
Novel TE Subfamilies in Rice STCs
Despite the over-representation of retroelements in the rice STCs, the current theoretical density of 1 STC every 9 kb across the rice genome affords us many possibilities to observe STCs homologous to TEs unknown previously or rarely discovered in rice. We found STCs homologous to maize Activator, En/Spm, andMutator transposons as well as Mariner transposons and pararetrovirus coat proteins. Phylogenetic analyses of these sequences revealed two separate subfamilies of Activator, several subfamilies of Mariner paralogs in various plants, and a potentially novel endogenous pararetrovirus in rice.
Activator
We found 75 STCs with homology to maize Ac ORF1, but no STCs homologous to Ac ORF2. A Fitch-Margoliash (1967) protein phylogeny of Activator ORF1 sequences, including two riceActivator homologs identified in the STC database, showed two separate paralogs of Activator present in rice (Fig.3A). Rice STC OSJNBa0076F14f is probably a rice ortholog of Activator, because the branching pattern of maize, pearl millet, and rice is the same as would be expected from a species phylogeny (Macrae et al. 1990, 1994; Paterson et al. 1996). Clearly, the rice STC OSJNBa0005B04f is a paralog of Activator and may have diverged from the line leading to Activator and snapdragon Tam3 early in plant evolution.
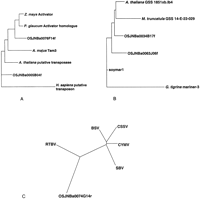
Phylogenies of TE homologs in the rice STC database. All phylogenies were constructed using the Fitch-Margoliash (1967) method. (A) Phylogeny of Activator-like protein sequences, derived from a partial-length multiple sequence alignment of 197 amino acids. Sequences from top to bottom are maizeActivator (P08770), pearl millet Activator homolog (1091678), rice STC OSJNBa0076F14f, snap dragon Tam3 (S13518),Arabidopsis putative transposase (AAD24567), rice STC (OSJNBa0005B04f), and human putative transposon (NP_004720). Translations of rice STCs were obtained from TFASTX alignments of maizeActivator (P08770) with the STC database. (B) Phylogeny of Mariner-like protein sequences, derived from a partial-length multiple sequence alignment of 107 amino acids. Sequences from top to bottom are Arabidopsis thaliana genome survey sequence 1851xb.lb4 (AF005799),Medicago truncatula genome survey sequence 14-E-22–029 from the Crop Biotechnology Center, Texas A & M University (AQ841462), rice STCs OSJNBa0034B17f and OSJNBa0063J06f, soybean Marinerelement soymar1 (AAC28384), and flatworm Girardia tigrina mariner-3 (CAA56859). Translations of rice STCs and other genome survey sequences were obtained from TFASTX alignments ofsoymar1 with the rice STC database and GenBank. (C) Phylogeny of pararetrovirus coat protein sequences, derived from a partial-length multiple sequence alignment of 220 amino acids. Sequences are from rice tungro bacilliform virus (RTBV, AAD30194), banana streak virus (BSV, CAA05264), cacao swollen shoot virus (CSSV, AAA03171), Commelina yellow mottle virus (CYMV, S11479), sugarcane bacilliform virus (SBV, S27938), and rice STC OSJNBa0074G14r. Translation of rice STC was obtained from a TFASTX alignment with CYMV protein 3 (S11479).
En-Spm/Tam1
We found 324 STCs homologous to the TNP2 protein fromAntirrhinum TAM1 transposon (CAA40555), making it the most abundant class II transposon in the STC database. Over-representation could occur, as TNP2 is 752 amino acids, and multiple STCs from the same genomic element may align to different regions of the TNP2 query. Nevertheless, the large quantity of TNP2 homologs implies that rice genome contains a substantial amount of En-Spm/Tam1-like transposons, even though no activity of En/Spm elements has been detected in rice so far.
Mutator
A total of 122 STCs were found to be homologous to the maizemudrA gene product, suggesting that the rice genome may contain Mutator-like elements; however, the most similar STC (OSJNBa0036C06f) is only 55.8% identical in a 238-amino acid alignment. The previously known rice mudrA homologOs-MuDR (AB012392, Yoshida et al. 1998) is also not present in our STC database (the closest match is only 47.5% identical over a 120-amino acid alignment). Together, these results imply the presence of a number of mudrA paralogs in the rice genome.
Mariner
Five STCs were identified as homologous to the soybeanmariner-like transposon soymar1 (AAC28384). A Fitch-Margoliash protein phylogeny of translations of these STC sequences together with other plant mariner homologs identified from GenBank reveals that the rice STCs are probably not orthologous to soymar1 (Fig. 3B). From the phylogeny, it appears that soymar1 and the other plant mariner-like elements diverged early in plant evolution. A minimum of twomariner paralogs appear in the rice STCs alone, and, if they are orthologous to each other, the Arabidopsis andMedicago genome survey sequences shown in the phylogeny comprise a fourth plant paralog of Mariner. During the preparation of this work, several mariner-like sequences have been identified and annotated in rice genomic sequences (AF172282,AP000837, AP000836); although to our knowledge, this is only the second published report of a monocot mariner homolog (Tarchini et al. 2000).
Pararetrovirus coat proteins
Although technically not TEs, fragments of a unique pararetrovirus sequence found in the tobacco genome (TPVL) interspersed at an estimated frequency of 103 per diploid genome (Jakowisch et al 1999). Jakowisch et al. suggest that a special mechanism of pararetrovirus dispersion and integration is sustaining such an unusually high copy number in the tobacco genome. To assess whether similar pararetrovirus-like sequences exist in the rice genome, we compared 36 pararetrovirus protein sequences with the rice STC database using TFASTX. The results showed that only three STCs are homologous to a pararetrovirus coat protein sequence found in Commelina yellow mottle virus, rice tungro bacilliform virus, and banana streak virus. Further, a multiple sequence alignment (data not shown) revealed that these three were most likely from the same element that integrated at minimum three times in the genome. The very low frequency of these homologs suggests that pararetrovirus-like sequences, such as TPVL, are not present in the rice genome; however, a Fitch-Margoliash protein phylogeny of these coat proteins (Fig. 3C) shows that the rice STC sequence is most similar to the coat protein sequence from rice tungro bacilliform virus but is not identical. This divergence may have resulted from a very ancient integration of the protein sequence of the tungro bacilliform virus, or the existence of an unknown rice pararetrovirus that is distantly related to the tungro bacilliform virus.
Miniature Inverted-repeat Transposable Elements
The first reported MITEs were the maize Tourist andStowaway families (Bureau and Wessler 1992, 1994a,b), which were subsequently reported in rice (Bureau et al 1996; Song et al. 1998). To identify MITEs in the rice STC database, a FASTA search (Pearson and Lipman 1988) was performed against the STC database by use of 23 known MITEs as query sequences (Bureau et al. 1996; Song et al. 1998). Because DNA—DNA sequence comparisons detect distant homology relationships poorly (States et al 1991; Pearson 1997), the sequence of the lowest-scoring significant STC with a full-length alignment to a known MITE was also used as a query in a second FASTA search of the rice STC database. Even so, the total number of MITEs was almost certainly underestimated and should be considered as a minimum only.
A total of 2746 STCs were found to contain various MITES as shown in Table 2. Several rice MITEs were represented abundantly, with nine MITEs showing homology to >100 STCs. The most abundant MITE in the rice STC database is Truncator, with 491 unique homologous STCs, followed by Tourist with 391 homologs, and Wanderer with 353 homologs. The two least frequent MITEs in the STC database are Krispie (no STC homolog) andPop (11 STCs). Interestingly, apart from maizeTourist and Stowaway, no non-rice MITEs were present in our STC database. Searches with bell pepper Alien (X87869),Medicago Bigfoot (AJ237732), maize Heartbreaker(transcribed from Zhang et al. 2000), and sorghum S-1,S-2, and S-3 (annotated in AF010283) showed no homologous STCs. Furthermore, MITEs that were first discovered in African Oryza species (Crackle, Krispie,Pop, and Snap from O. longistaminata andp-SINE1 from O. glaberrima) appear to occur with less frequency than other rice MITEs. Whereas known Oryza sativaMITEs occur with an average number of 222.6, non-sativa MITE occur with an average number of only 15. The lack of most of the non-rice MITEs and the biased representation of non-sativaMITEs in the STC database strongly supports a species-specific distribution for MITEs.
MITEs Identified in the STC Database by FASTA Searches
Bureau and Wessler (1994a) have noted that the MITE Touristappears to be associated with genes in maize, rice, and sorghum; however, their sample size was very low. Recent work on the maizeHeartbreaker element confirms that these MITEs also appear to be associated with genes (Zhang et al. 2000). To ascertain whether this positional bias of MITEs extends to all MITEs in the rice genome, we used BLASTN (Altschul et al. 1997) to compare the rice STC library with the TIGR Rice Gene Index (OGI; Quackenbush et al. 2000). Our results show that 48.3% of MITE-containing STCs (MITE–STCs) are also homologous to a sequence in OGI (BLASTN E<10−7); whereas only 11.5% of MITE-lacking STCs show homology to an OGI sequence. This bias is more remarkable when one considers the average length of the STCs; when an STC shows homology to both an OGI and a MITE sequence, the MITE must be within only a few hundred nucleotides of the transcription region.
Broken down by MITE, we find a surprising variation of gene positioning among the different MITE families (Fig. 4). Only 10.5% of 181 Explorer-containing STCs are also homologous to an OGI sequence, but nearly every Stowaway-containing STC (95.8% of 166) is also homologous to an OGI sequence. It is impossible to say whether our results indicate that certain MITEs do not insert near genes in the rice genome or that some MITEs insert further than a few hundred nucleotides from the transcription region. In either case, our results clearly demonstrated that the association of MITEs with genes is not uniform among different MITEs.
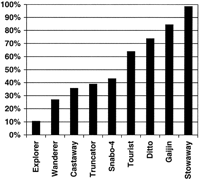
MITEs are differentially associated with ESTs. Percentage of MITE-containing rice STCs that also show homology to a sequence in the Rice Gene Index (BLASTN E-value<10−6), displayed by MITE type. Only MITEs with >100 STC homologs are shown.
Rice TEs Are Not Clustered
TEs in plants with small genomes such as Arabidopsis(∼130 Mb) were shown clustered only at the pericentromeric regions (Lin et al. 1999; Mayer et al. 1999). Similarly, Ty3/Gypsy-related DNA fragment from sorghum has been shown present in centromeres of sorghum, wheat, maize, and rye (Miller et al. 1998), and several centromeric repeats from the rice cultivar Indica are also retroelement-related (Dong et al. 1998). On the other hand, in grasses with large genomes such as maize (∼2500 Mb), retrotransposons can be clustered along the chromosomes, inserting between the genes (SanMiguel et al. 1996, 1998). Recent work has shown that the large size of maize genome is largely due to retroelements that have inserted in the last 6 million years (SanMiguel et al. 1998). To analyze possible positional bias of TEs in the rice genome, we mapped our TE–STCs onto the physical map contigs assembled at CUGI. Presently, the CUGI physical map consists of 73,728 clones in 1018 contigs (G. Presting and R. Wing, unpubl.). To estimate gene location, we have mapped EST-containing STCs to this map as well.
We identified EST matches using BLASTN to search the rice gene index (OGI) as described above. STC matches from both the OGI and TE database searches were associated with their physical contigs, and the TE and EST contents of each contig were examined. If TEs were positioned in the rice genome away from genes, we would expect to see a negative correlation between TE and EST content of the physical map contigs, but our results show no correlation whatsoever (Fig. 5). This implies that the TEs and genes of the rice genome appear to be randomly distributed.
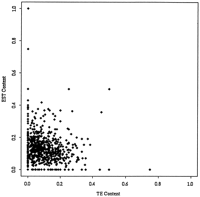
A scatterplot of the EST and TE contents on 1018 rice contigs. TE homologs in the STC database were identified by FASTX searches (E<10−5), as described in text. EST homologs in the STC database were identified by BLASTN searches of the Rice gene index (E<10−6) using STCs as queries.
DISCUSSION
The TE Compositions in the Rice Genome
We analyzed the TE content in 73,362 STC sequences by a protein homology search of each STC against a set of 1358 TE proteins downloaded from GenBank. A total of 6848 STCs were found to contain regions homologous to the known TEs, representing 9.3% of the STCs in the rice STC database. In contrast to a survey of the TEs on a 1-Mb PAC contig from chromosome 1, our TE–STCs were primarily retroelements (88.0%). The TEs on the 1-Mb PAC contig were only 66.1% retrotransposon. The over-representation of retrotransposons in the rice STCs is not due to the redundancy of the rice database, and curiously enough, is also observable in 16,360 ArabidopsisSTCs. Nevertheless, counting MITE, retrotransposon, and transposon alignments with the redundant STCs, we find that the TE–STCs discussed in this paper cover 2.2 Mb of genomic DNA, only 4.5% of the total sequenced nucleotides. Although the actual number of TEs will remain unclear until the whole rice genome is sequenced, our present analysis shows that TE content of the rice genome is probably <10%.
Our FASTX survey of the rice STCs also revealed that almost all known TEs are present in the rice genome. Sequences of 821 STCs were homologous to class II TEs, including maize Activator,En/Spm, and Mutator. Transposons that are rarely known in plants, such as mariner, were also present in the STC database. Phylogenetic analyses of the mariner elements identified in this study reveal the existence of multiple subfamilies of mariner in plants. We also identified what appears to be a novel variety of rice tungro bacilliform virus, which appears to be endogenous to the rice genome.
Our results also show the abundance of MITEs in the rice STC database. We found 2746 STCs that contain regions homologous to known MITEs. Some MITEs, such as maize Stowaway, are found in numerous species of plants, including both monocots and dicots (Bureau and Wessler 1994b), but our results clearly show a species-specific distribution of many MITE sequences. MITEs first identified in African rice species are present in only low copy numbers in the Nipponbare STC database. Furthermore, we also showed that the gene-preferring insertion bias of some MITEs may not be universal to all MITEs. Although bothExplorer and Stowaway MITEs were found in >100 STCs, only 10.5% of Explorer-containing STCs compared with 98.5% of Stowaway-containing STCs were found to also contain regions homologous to a sequence in the rice gene index, indicating the presence of a gene. This difference may be due to true insertion bias of Explorer and Stowaway, positional bias (Explorer inserts near genes but far enough from the transcript to be undetectable in the STC database), or a representation bias in the rice gene index (Explorer inserts near genes that are transcribed infrequently and thus unlikely to be detected in an EST survey). In any case, our results clearly show the usefulness of MITEs for gene discovery as nearly half (48.3%) of the MITEs identified in the STC database were within a few hundred nucleotides from transcription regions. MITEs may be especially important for crop plants with large genomes, such as maize, barley, and wheat, for which no large-scale genome-sequencing project will be attempted in the near future.
The Distribution of TE–STCs Across Rice Genome and Implications for Genome Sequencing
The completion of two Arabidopsis chromosomes (2 and 4) for the first time provides insight into the physical distribution of TEs along higher plant chromosomes (Lin et al. 1999; Mayer et al. 1999).Arabidopsis TEs are mainly clustered around the centromeres. Clusters of retrotransposons have been reported in the intergenic regions on the maize chromosomes where retrotransposons constitute up to 50% of the genome (SanMiguel et al. 1996). Although 340 kb of genomic DNA surrounding the Adh1 gene from rice has been analyzed, the insertion of large clusters of retroelements was not observed in the rice intergenic regions (Tarchini et al. 2000). Our analysis of the physical location of 6848 TE–STCs did not reveal obvious TE clustering regions in 1018 physical map contigs, confirming the results ofTarchini et al. (2000).
The STC strategy to identify a minimum tile of large-insert clones for genome sequencing has been applied to the human andArabidopsis genome projects (Venter et al. 1996) and has proven to be highly effective (Kelley et al. 1999; Siegel et al. 1999). The low content of TEs in the STC database and their apparent random distribution on the physical map both confirm the quality of the rice genome as a model crop genome. The lack of large blocks of known retrotransposons, which require painstaking effort to resolve during sequence assembly, is good news for the rice genome sequencing community. With the international rice genome project now on track, a complete assay of the sequence composition and organization of rice genome will soon become reality and will provide a more lucid picture of the role of transposable elements in the genome evolution of rice and related cereals.
METHODS
BAC End Sequencing
A total of 4 μl of BAC culture in LB freezing medium was inoculated into 4 ml of LB medium containing chloramphenicol and incubated for 20 hr at 37°C. BAC DNA was isolated using the Autogen 740 (Integrated Separation System) according to the manufacturer's instructions. DNA pellets were resuspended in 25 μl of 1 mm Tris.HCl (pH 7.5). A total of 20 μl were used as the template for sequencing reactions in a total volume of 30 μl (5 μl of ABI Big Dye (Perkin Elmer); 50 pmole primer; 1.75 μl sequencing buffer containing 800 mm Tris.HCl (pH 9.0) and 20 mm MgCl2; 2.25 μl dH2O). Cycle sequencing reactions were performed as one cycle for 4 min at 95°C, followed by 70 cycles of 15 sec at 95°C, 10 sec at 51°C, and 4 min at 60°C. Cycle-sequencing products were precipitated with ethanol containing 1/3 volume of 7.5 m NH4OAc and run on ABI377 automatic sequencers. The sequence traces were then transferred to a Sun workstation and base called by Phred, and vector sequences were masked by CROSS_MATCH software packages (Ewing and Green 1998).
Sequence and Statistical Analysis
FASTX (Pearson et al 1997) was used to compare all Nipponbare STCs with a database of 1358 transposable-element protein sequences obtained from GenBank, by use of batch Entrez. Additional transposable elements were detected by FASTA searches (Pearson and Lipman 1988) of the STC database using known MITEs as queries and by TFASTX (Pearson et al. 1997) searches using pararetrovirus protein sequences as queries. For phylogenetic analysis, CLUSTALW (Thompson et al 1994) was used to generate multiple sequence alignments, and the PROTDIST and FITCH programs of the PHYLIP package (Felsenstein 1993) were used to estimate sequence distances and phylogenies, respectively. For all alignments used in phylogenies, translations of the STCs were derived from FASTX alignments and end gaps were trimmed. Statistics were calculated using Splus version 5. All FASTA, FASTX, and TFASTX searches were run on a Dell PowerEdge2300 server running LINUX 6.1; all other software were run on a Sun Ultra30 running Solaris 2.6. The complete CUGI STC database is available at ftp.genome.clemson.edu.
Acknowledgments
We thank the staff of the CUGI BAC/EST Resource, Sequencing, Physical Mapping, and Bioinformatics Centers for supplying the resources and generating and processing the sequence data used for this analysis. We especially thank Dr. P. San Miguel for sharing insights on cereal transposable elements and his critical reading of an earlier version of the manuscript and Mr. R. Kingsburry III for his help with initial computer analyses. This work was funded in part by grants from Novartis, NSF-MRI # 9724557 to R.A.W. and R.A.D., NSF Plant Genome # DBI-987276 to R.A.W., R.A.D., M.S., and D.F., and the Rockefeller Foundation RF98001#630 and the Coker Endowed Chair to R.A.W. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding agencies.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Present addresses: 3Orion Genomics, St. Louis, Missouri 63108 USA; 4Department of Agronomy, Konkuk University, Seoul, South Korea 143-701, Korea; 5Institute for Computational Genomics, 110 Clemson, South Carolina 29631 USA; 6Department of Plant Pathology, North Carolina State University, Raleigh, North Carolina 27606 USA.
-
↵7 Corresponding author.
-
E-MAIL rwing{at}clemson.edu; FAX (864) 656–4293.
-
- Received January 14, 2000.
- Accepted May 17, 2000.
- Cold Spring Harbor Laboratory Press








