
Tandem Clusters of Membrane Proteins in Complete Genome Sequences
Abstract
The distribution of genes coding for membrane proteins was investigated in 16 complete genomes: 4 archaea, 11 bacteria, and 1 eukaryote. Membrane proteins were identified by our new method of predicting transmembrane segments (Kihara et al. 1998) after the removal of amino-terminal signal peptides. Interestingly, about half of the membrane protein genes in each genome were found to be located next to another, forming tandem clusters. Roughly 10%–30% of the tandem clusters were conserved among organisms, and most of the conserved tandem clusters belonged to one of the three functional groups, namely, transporters, the electron transport system, and cell motility. A tandem cluster sometimes contained paralogous membrane proteins, in which case the cluster size and the number of transmembrane segments could be related to a functional category, especially to transporters. In addition to the clustering of membrane proteins, the clustering of membrane proteins and ATP-binding proteins in the complete genomes was also analyzed. Although this clustering was not statistically significant, it was useful to identify candidate membrane protein partners of isolated ATP-binding protein components in the ABC transporters. Possible implications of tandem cluster organization of membrane protein genes are discussed including the complex formation and other functional coupling of protein products and the mechanism of protein translocation to the cell membrane.
With the wealth of complete genome sequences accumulated by the recent genome projects, we now have the opportunity to analyze genome structure and function comprehensively from the catalog of all the genes encoded in the genome. It is possible, for example, to compare the ordering of genes in different genomes and to understand general principles of how functionally coupled genes are physically encoded in the genome and possibly coregulated at the level of gene expression. The correlation of functional coupling and physical coupling seems to be prevalent in bacterial and archaeal genomes; namely, a set of functionally correlated genes tends to be encoded in a potential operon (Tamames et al. 1997; Dandekar et al. 1998; Overbeek et al. 1999). Thus, the analysis of conserved gene orders among different genomes provides significant clues to functional annotation of individual genes, as additional information to conserved sequence similarity. Furthermore, the prediction of higher order structures may be utilized in order to compensate for the limitation of the sequence similarity search for functional identification. Aurora and Rose (1998)used predicted secondary structures in the search for a particular enzyme and Fetrow and Skolnick (1998) created templates of active sites of enzymes suitable for the screening of genome sequences.
We combine the prediction of membrane proteins with the analysis of gene orders and sequence similarity in the complete genome sequences. Membrane proteins play important roles in living cells, such as for transport, energy production, and cell signaling. Previous studies on membrane proteins in comparative analysis of genome sequences were concerned mostly with the estimation of the number of membrane proteins (Arkin et al. 1997; Boyd et al. 1998; Jones 1998; Wallin and von Heijne 1998). Paulsen et al. (1998) analyzed a specific class of membrane proteins, namely transporters, and discussed the gene distribution in the genome in relation to the environment in which each organism inhabits. Tomii and Kanehisa (1998) also performed a systematic survey of ABC transporters and operon structures in the complete genome sequences, although their analysis did not make use of predictions of transmembrane segments.
Recently, we have developed a new prediction method of membrane proteins, which takes into account the number and the types of transmembrane segments (Kihara et al. 1998). Here, membrane proteins are detected by our prediction method together with the method to remove amino-terminal signal peptides, which are often misidentified as transmembrane segments of mature proteins. It is found that a surprising portion of membrane proteins are encoded as gene clusters, and the pattern of conservation can be used for the prediction of functional categories of membrane proteins. We further report the clustering of membrane proteins and ATP-binding proteins in the genome, which is not statistically significant, but which contains a major class of membrane protein machinery—ABC transporters.
RESULTS
Estimated Amounts of Membrane Proteins
First, we analyzed the amount of membrane proteins in the complete genome sequences of 16 organisms. The number of predicted membrane proteins is shown in Table 1. The proportion of membrane proteins in each organism ranges from 18%–29%. This estimate is smaller than that based on a transmembrane prediction method only, which reports the values ∼35% (Frishman and Mewes 1997). The discrepancy may be attributable to the removal of amino-terminal signal peptides in our analysis. The estimates forHaemophilus influenzae and Escherichia coli are almost the same as those by Jones (1998) who also masked out the signal peptides in the preprocessing step; however, his value forSacchromyces cerevisiae, 18%, is much smaller than ours.
The Numbers of Predicted Membrane Proteins and ATP-Binding Proteins
The number of probable genes or open reading frames (ORFs) is known to be roughly proportional to the genome size in bacteria and archaea where the gene (ORF) density is about one per 1,000 bases (Table 1). This is to be compared with one per 2,000 bases in S. cerevisiae and one per 5,000 bases in Caenorhabditis elegans. There is a tendency that the proportion of membrane proteins increases with the number of ORFs in the genome (Wallin and von Heijne 1998); membrane proteins are relatively more abundant in a larger genome, which is also observed in Table 1. Generally speaking, facultative bacteria tend to have larger genomes than obligate bacteria, which are correlated with higher proportions of paralogous genes. Thus, we estimated the proportions of paralogous proteins separately for membrane proteins and nonmembrane proteins. The result is shown in Figure 1. First, paralogs are in fact more abundant in larger genomes, which are clustered in the upper righthand corner of Figure 1A or in the upper portion of Figure 1B. Second, the proportion of paralogous proteins increase with the number of proteins in the pool and seem to become saturated at ∼55% (Fig.1B). Third, when the numbers of proteins are compared in different species, membrane proteins generally contain higher proportions of paralogs than nonmembrane proteins (x vs. ● in Fig.1B). The additional repertoire of membrane proteins is likely to be used to generate functional diversity and to cope with varying environmental factors.
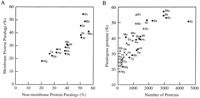
(A) The proportion of paralogous proteins is plotted for membrane proteins vs. nonmembrane proteins in 16 organisms (see Table 1for abbreviations). A paralog is defined by the Smith-Waterman score of 150 or more by SSEARCH after preprocessing with SEG. (B) The proportion of paralogous proteins is plotted against the total number of membrane or nonmembrane proteins in 16 organisms. (x) Membrane proteins, (•) nonmembrane proteins.
The distribution of membrane proteins grouped by the number of transmembrane segments was very similar to those reported previously (Arkin et al. 1997; Jones 1998; Wallin and von Heijne 1998; data not shown). Membrane proteins with three or more predicted transmembrane segments are highly likely to be true membrane proteins (see Discussion). They constitute roughly half of all the membrane proteins in our analysis (36%–60% in Table 1).
The Tandem Clusters of Membrane Proteins
Next, we investigated the gene distribution pattern of membrane proteins in the complete genome sequences. Surprisingly, in all 16 organisms ∼50%of the membrane proteins were found to be located next to each other, namely, in tandem clusters. Table2 shows the number of membrane proteins in tandem clusters, as well as the number of tandem clusters of membrane proteins in each genome. The statistical significance of the number of membrane proteins in tandem clusters, which we call here the score, was estimated by randomizing the locations of all the ORFs in the genome. For each organism, the randomization was performed 1000 times and the mean and the standard deviation of the scores were calculated. The actual score can then be converted to the Z value (Table 2), which is the number of standard deviation units from the mean. Assuming that the score follows the normal distribution, the probability of observing the actual score by chance alone can be determined. The probability was <0.03%, except for Treponema pallidum (4.6%) andPyrococcus horikoshii (2.7%).
The Number of Membrane Proteins in Tandem Clusters
We have defined a tandem cluster simply as a group of adjacent membrane protein genes in the genome. However, in most of the
cases a tandem cluster is formed by the genes encoded on the same strand (Table 2, fourth column) and, furthermore, the gaps between genes are usually <300 bp, which is the condition used by Overbeek et al. (1999) to define a gene cluster (Table 2, fifth column). Thus, it is possible that most of the tandem clusters correspond to operon structures, except for S. cerevisiae. In
 of the cases, a tandem cluster spans both strands although it is not clear whether it is under the same gene regulatory mechanism.
About 60%–80 % of the tandem clusters are of size two. The longest cluster is found inMethanobacterium thermoautotrophicum, containing 12 genes on the same strand, MTH384–MTH395, which corresponds to 10 functionally unknown proteins and 2 subunits
of NADH dehydrogenase.
of the cases, a tandem cluster spans both strands although it is not clear whether it is under the same gene regulatory mechanism.
About 60%–80 % of the tandem clusters are of size two. The longest cluster is found inMethanobacterium thermoautotrophicum, containing 12 genes on the same strand, MTH384–MTH395, which corresponds to 10 functionally unknown proteins and 2 subunits
of NADH dehydrogenase.
Conserved Tandem Clusters
When the sequence similarity of constituent membrane proteins was examined, some of the tandem clusters were conserved between organisms and/or within an organism. The proportion of such conserved tandem clusters is shown in the last column of Table 2. It was in the range of 10%–30% in bacteria and archaea, but it was 2% in S. cerevisiae. The conserved clusters almost exclusively (97.6%) consisted of membrane protein genes encoded on the same strand with gaps of <300 bp, most likely representing conserved operons.
The majority (97.3%) of the conserved tandem clusters could be associated with known functions, which are summarized in Table3. They belong to one of the three functional categories: membrane transporters, the electron transport system, and cell motility. A large fraction of the transporter category was formed by the ABC (ATP-binding cassette) transporters (Higgins 1992; Fath and Kolter 1993; Dean and Allikmets 1995). The conserved operon structures of ABC transporters are known to be related to the grouping of substrate specificity (Saurin and Daussa 1994; Tomii and Kanehisa 1998), which is also observed in Table 3. The average cluster size for the transporters is small, ∼2.6. In contrast, the electron transport system consists of a larger cluster size, ∼5, and the constituent membrane proteins are also larger, with >10 transmembrane segments. In many cases the membrane proteins encoded in a conserved tandem cluster, i.e., in a conserved operon, are likely to interact physically (Dandekar et al. 1998)— two permease proteins forming a channel for the ABC transporter, multiple subunits forming an enzyme complex, or multiple subunits responsible for chemotaxis and flagellar assembly. In the last category of other functions in Table 3, rod shape-determinant protein (RodA) and penicillin-binding protein 2 (Pbp2) are responsible for the cell wall formation; RodA activates Pbp2, which synthesizes peptideglycan.
Conserved Tandem Clusters with Known Functions
In addition to the conserved clusters shown in Table 3, there were eight conserved clusters consisting of hypothetical membrane proteins as shown in Table 4. Two of them are conserved among three organisms; cluster no. 1 is conserved in E. coli,H. influenzae, and Bacillus subtilis and cluster no. 2 is conserved in Methanococcus jannaschii, Archaeoglobus fulgidus, and P. horikoshii.
Conserved Tandem Clusters with Unknown Functions
As mentioned above, S. cerevisiae has a very low rate of conservation of tandem clusters, and all the eight conserved clusters of S. cerevisiae (Table 2) are conserved only within the organism. This is consistent with the fact that S. cerevisiaedoesn't have bacteria-like operons (Zhang and Smith 1998). For example, the genes for the subunits of cytochrome c oxidase and ATP synthase are scattered in different chromosomes.
Gene Duplication in Tandem Clusters
In the above analysis of conserved clusters, the sequence similarity of membrane proteins was used to define the similarity relationship between two tandem cluster units. Here the sequence similarity is examined within a tandem cluster unit to identify possible gene duplications of constituent membrane proteins. More than 10% of the tandem clusters in each organism are found to contain paralogs, i.e., pairs of constituent membrane proteins that are similar to each other. In Figure 2 the number of such membrane proteins with similar partners is compiled against the size of the belonging clusters and the predicted number of transmembrane segments. To correlate with functional information, the compilation is made separately for three groups: ABC transporters, other transporters, and the rest. The membrane proteins of ABC transporters (Fig. 2A) exhibit the most characteristic features. The cluster size is two in most cases and the number of transmembrane segments peaks at around six and seven (Higgins 1992; Tam and Saier 1993). At the same time there are significant variations of both the number of transmembrane segments and the number of membrane proteins in an operon. The maltose transporter (MalF) is experimentally known to contain eight transmembrane segments (Froshauer et al. 1988), but the number of predicted transmembrane segments can vary more drastically as seen in Figure 2A.
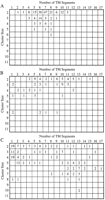
The frequency of membrane proteins that are paralogous within a tandem cluster, where each membrane protein is classified according to the number of transmembrane segments and the size of the cluster to which it belongs. The criterion for a paralog is the same as in Figure 1. The frequency is counted separately for three functional groups: (A) ABC transporters; (B) transporters other than ABC transporters; and (C) membrane proteins of other functions. In total, 256 clusters (13.0%) contained such pairs of paralogous membrane proteins, and among them, 98 (38.3%) were those of ABC transporters, 37 (14.5%) were those of other transporters, 63 (24.6%) were those of the other functions, and 58 (22.7%) were those of hypothetical functions (not shown).
The membrane proteins for the transporters other than the ABC transporters are shown in Figure 2B. The cluster sizes of cation ATPases are three in most cases. Membrane proteins with 10 predicted transmembrane segments and in the cluster of size three are sodium- and calcium-transport ATPases. Clusters of larger sizes (>3) are a mixture of membrane proteins of various kinds, though some of them are still hypothetical proteins and thus their functions could not be assigned. Membrane proteins with clusters of other functions (Fig. 2C) are clearly differentiated into two groups: those with a larger cluster size (∼6) and many transmembrane segments (∼12) and those with a smaller size (∼3) and a few transmembrane segments (1 or 2). The former group corresponds to the complexes of membrane proteins in the respiratory chain, namely, NADH dehydrogenase and cytochrome c oxidase. The latter group corresponds to various kinds of membrane proteins, including peptide synthase, kinase, methyl-accepting chemotaxis protein, surfactin synthase, and others.
The observation made here may be used for functional assignment of membrane proteins without any sequence similarity to known proteins. In fact, a simple rule of discriminating transporters can be established according to the frequencies of transporters and nontransporters. When a tandem cluster of membrane proteins contains paralogs, and if the number of predicted transmembrane segments and the size of the cluster are in the range shown in Figure 3, then the cluster is likely to be a transporter. A darker box in Figure 3 represents higher likelihood, which is defined in three levels according to the relative frequency and the absolute number of transporters in Figure 2. Level 1 corresponds to the relative frequency of 0.85 or higher with ⋝12 observed instances. Levels 2 and 3 correspond to the relative frequency of 0.8 or higher with ⋝5 and 3 instances, respectively. Although this empirical rule is derived from a small number of samples, we believe that it is still useful to obtain any functional clue to the large number of genes left unassigned in the completely sequenced genomes. In our data set there were 55 tandem clusters containing paralogs whose functions were not known. Based on this empirical rule we predict seven transporters which are shown in Table 5.

When a tandem cluster of membrane proteins contains paralogs, the cluster size and the numbers of transmembrane segments in paralogous membrane proteins may be used to assign functions. Here the likelihood of being a transporter (A or B in Fig. 2) rather than in another functional category (C in Fig. 2) is shown by the darkness in three levels, darker meaning more likely. The likelihood is defined by the relative frequency and the absolute number of transporters in Figure 2 (see text for details).
Predicted Transporters According to the Number of Transmembrane Segments and the Cluster Size
Clustering of Membrane Proteins and ATP-binding Proteins
Because ATP-binding proteins provide energy for active membrane transport and other cellular machineries, we suspected that
there would be a tendency for membrane proteins and ATP-binding proteins to form clusters in the genome. This is certainly
the case for the ABC transporters, whose operons generally contain adjacent permease proteins and ATP-binding proteins. In
the 16 complete genomes, ATP-binding proteins constituted between 5% and 13% of ORFs (Table1). We defined a cluster for each ATP-binding protein together with all the membrane proteins and ATP-binding proteins within
five gene positions on both sides. When such physical coupling of membrane proteins and ATP-binding proteins was searched,
roughly 21%–39% of ATP-binding proteins were adjacent to membrane proteins. However, this coupling was not statistically significant
(data not shown). On average, ∼
 of all the pairs of membrane proteins and ATP-binding proteins are conserved among different organisms as shown in Table 6. More than one-half of the conserved pairs fall in the category of ABC transporters. In addition, the conserved pairs include
protein-export proteins, proteins involved in twitching motility, flagellar biosynthesis or secretion, pairs of gluconokinase
and gluconate transporter, V-type ATP synthases, polyketide synthases, acriflavin resistance proteins, sporulation proteins,
pairs of signal recognition particle protein, and protein-export membrane protein. Clusters of unknown functions were also
detected, but they were conserved only between two closely related species.
of all the pairs of membrane proteins and ATP-binding proteins are conserved among different organisms as shown in Table 6. More than one-half of the conserved pairs fall in the category of ABC transporters. In addition, the conserved pairs include
protein-export proteins, proteins involved in twitching motility, flagellar biosynthesis or secretion, pairs of gluconokinase
and gluconate transporter, V-type ATP synthases, polyketide synthases, acriflavin resistance proteins, sporulation proteins,
pairs of signal recognition particle protein, and protein-export membrane protein. Clusters of unknown functions were also
detected, but they were conserved only between two closely related species.
The Number of Adjacent Pairs of Membrane Proteins and ATP-binding Proteins
There was a relatively large group of conserved pairs within S. cerevisiae. This paralog group consists of five adjacent pairs, namely, (YML133C, YML132W), (YNL339C, YNL336W), (YHL050C, YHL048W), (YGR296W, YGR295C), and (YFL066C, YFL062W), respectively for the ATP-binding protein and the membrane protein. The membrane proteins are similar to subtelomerically encoded proteins and are predicted to have three transmembrane segments, except for YGR295C that contains just two. We believe that the physical coupling of these membrane proteins and the ATP-binding proteins has some functional relevance.
Prediction of ABC Transporter Components
The ABC transporters form the largest superfamily of paralogous proteins in bacterial and archaeal genomes (Tatusov et al. 1997;Paulsen et al. 1998). Typically, a transporter consists of three components: a pair of ATP-binding proteins, a pair of membrane proteins, and a substrate-binding protein. In bacteria and archaea the majority of these components are known to be located next to each other, probably forming operons (Tomii and Kanehisa 1998), but there are also isolated components. We have searched by sequence similarity such isolated components of ABC transporters in the complete genomes (Table 7) and tried to identify their partners (Table8). An ABC-transporter component was considered to be isolated when there was no other component within five gene positions on both sides. Note that the search was performed using the annotated set of bacteria-type ABC transporters in KEGG (Tomii and Kanehisa 1998). Therefore, many eukaryotic ABC transporters (Fath and Kolter 1993) in S. cerevisiae were not detected.
The Number of Bacteria-type ABC Transporter Components
Predicted Membrane Protein Components of ABC Transporters
As shown in Table 7, the degree of isolation depends on the organism; in Synechosystis and Aquifex aeolicus ∼40% of the components are isolated. Table 7 also indicates that ATP-binding protein components are more likely to be isolated than membrane protein components. However, this may simply be due to the fact that because membrane protein components are less conserved than ATP-binding protein components (Tomii and Kanehisa 1998), they have not been detected by sequence similarity searches. Candidates of missing membrane protein components, which are the partners of isolated ATP-binding protein components, may then be found by examining conserved pairs of ATP-binding proteins and membrane proteins. Table 8 summarizes the results of searching for missing membrane protein components. The newly identified pairs of the ATP-binding protein and membrane protein components are predicted to form new types of ABC transporters. Note that the maximum distance of two genes in a cluster is 10 gene positions, which is the case for HI1252 and HI1242 in cluster no. 1 in Table 8. They are in the same cluster that has HI1247 as its center, and because HI1247 is not a component of an ABC transporter, both HI1252 and HI1242 were termed to be isolated in Table 7.
Table 7 also shows the number of fused components. The majority of fused components belong to the class of multidrug-resistance family transporters (Tomii and Kanehisa 1998) where the membrane protein component is fused with the ATP-binding protein component. Occasionally two fused components are encoded as tandem repeats in the genome. InS. cerevisiae, most of the components are fused and isolated, but bacteria-type single domain components are also found.
DISCUSSION
We investigated the distribution of membrane proteins in 16 complete genome sequences. We showed that statistically significant portions of membrane proteins were encoded in the genome as tandem clusters. There was a total of 1957 tandem clusters in the 16 genomes (Table 2). We analyzed the sequence similarity of membrane proteins in tandem clusters in order to identify, first, conserved (orthologous and paralogous) tandem clusters and, second, paralogous proteins within a tandem cluster. Most of the conserved clusters and/or the clusters containing paralogs represented functionally well-identified proteins (Table 3 and Fig. 2). There were eight conserved clusters whose functions were not known (Table 4). We predicted seven transporters (Table 5) among 55 functionally unknown clusters containing paralogs, according to an empirical rule concerning the cluster size and the number of transmembrane segments. This was an attempt to use the information of structural features in functional annotation of membrane proteins without relying on sequence similarity. In addition, we identified probable membrane protein partners of isolated ATP-binding protein components in the ABC transporters by searching for adjacent pairs of membrane proteins and ATP-binding proteins.
Our analysis depends on the accuracy of predicting membrane proteins, for which we used the TSEG program (http://www.genome.ad.jp/SIT/tseg.html). As reported previously (Kihara et al. 1998), TSEG missed 14.9% of real transmembrane segments (false negatives) and overpredicted 8.5% of nontransmembrane segments (false positives) in our test data set. Let us assume that this prediction accuracy applies to the current genome-scale analysis and that every transmembrane segment is predicted independently with the above accuracy. Then the probability that a membrane protein predicted to have three transmembrane (TM) segments that are actually a globular (nonmembrane) protein is (0.085)3 = 6.1×10−4 and conversely, the probability that a real 3TM protein is predicted to be a globular protein is (0.149)3 = 3.3×10−3. Considering the number of predicted membrane proteins in the present analysis (Table 1), it is almost certain that those predicted to have three or more TM segments are real membrane proteins, and that real membrane proteins with three or more TM segments would not be missed. To take another example, the probability that a real 6TM protein is predicted to have three TM segments is 6C3×(0.149)3= 0.066 and the probability that a membrane protein predicted to have nine TM segments that are indeed a 6TM protein is 9C3×(0.085)3= 0.052. Though the probability depends on the predicted number of TM segments in a protein, these values provide an idea of how rare it is to mispredict the number of TM segments by three or more. With careful consideration of the limitation of the prediction accuracy, we think that the information of the predicted number of TM segments can be used in a positive way to understand protein functions.
The main conclusion of the present study is that about half of the membrane proteins form tandem clusters in the genome. There are several possible explanations for this observation and they are not necessarily mutually exclusive. First, the functional coupling of protein products is probably the most dominant biological constraint on such clustering in the genome. Despite the fact that the locations of orthologous genes are extensively shuffled even in the genomes of closely related species, some gene clusters are found to be tightly coupled as conserved operons (Tamames et al. 1997; Dandekar et al. 1998; Overbeek et al. 1999). The genome is viewed increasingly as a dynamic entity, and conserved gene clusters may also result from the horizontal gene transfer (Xu et al. 1998). Not including the conserved tandem clusters reported above, there may be other functionally coupled tandem clusters though they are not conserved among the organisms studied here. We expect that as more completely sequenced genomes become available the possibility of identifying functional clues will increase. Second, it is conceptually possible to imagine that the apparent clustering of membrane proteins results from the clustering of nonmembrane proteins as a background clustering. There are a number of known examples of functionally coupled nonmembrane protein clusters. However, nonmembrane proteins constitute 70%–80% of the total proteins, and the majority of nonmembrane proteins do not form gene clusters as evidenced by the extensive shuffling of orthologous genes. In fact, we believe that the functional coupling of protein products alone cannot explain the statistically significant occurrence of membrane protein gene clusters, although the functions of all membrane proteins are not yet known.
Third, we present a hypothesis that forming tandem clusters is favorable for the cellular mechanism of membrane protein expression, perhaps at the stage of protein translocation to the cell membrane. The bacterial protein translocation machinery is well studied inE. coli. One is the Sec machinery, which involves SecB that binds to the mature regions of nascent proteins and delivers them to SecY/E/G translocon, using the energy of ATP hydrolysis by SecA and proton motive force (Tokuda 1994). Another involves the signal recognition particle (SRP) that interacts with the hydrophobic signal peptide of a nascent protein. The two translocation pathways seem to use the common translocon (Valent et al. 1998). It has been shown that a subset of membrane proteins is dependent on the SRP pathway but others are not (Ulbrandt et al. 1997). We can speculate an implication of tandem clusters for the SecB machinery. Considering the report that SecB forms a tetramer and can bind more than one polypeptide chain (Randall et al. 1998) and also the fact that bacterial mRNA is usually polycistronic, it may be favorable for the genes of membrane proteins to be positioned tandemly, so that SecB delivers them all together like an omnibus. As for S. cerevisiae, we cannot reason in the same way because it does not have a SecB-like protein (Lyman and Schekman 1996), though the translocon complex is similar to that of bacteria (Jungnickel et al. 1994). Still, there may be some biological implications in S. cerevisiae as well, because tandem clusters of membrane proteins are as abundant as in bacteria (Table 2) without bacteria-type operon structures. Further experimental analysis of genome-scale translocation mechanism is required for the validation of our hypothesis.
Although the locations of genes and their amino acid sequences can be determined rapidly by whole genome sequencing, the functional identification of individual genes has been a slow and tedious process. We have shown that missing permease protein components of ABC transporters may be identified by searching for conserved clusters of membrane proteins and ATP-binding proteins. Generally speaking, this type of analysis extends the current knowledge on functions in terms of physical coupling of genes. Namely, when the function is known for only one of the two genes but the other gene is physically coupled, then the known function may be extended to include both genes. The analysis can be further generalized to include other types of couplings, such as identifying sequence motifs that are known to be present on two interacting proteins. Furthermore, new experimental methods in functional genomics provide direct information about coupling of genes; especially cDNA microarrays (Brown and Botstein 1999) at the level of mRNA expression and yeast two hybrid systems at the protein–protein interaction level. Thus, based on the concept of links, or binary relations (Kanehisa 2000), both computational predictions and data processing of systematic experiments can be integrated to identify functional couplings and eventually to understand the entire network of genes and proteins.
METHODS
Complete Genomes
We analyzed the complete genome sequences of the following 16 organisms: M. jannaschii (Bult et al. 1996), M. thermoautotrophicum (Smith et al. 1997), A. fulgidus(Klenk et al. 1997), and P. horikoshii (Kawarabayasi et al. 1998) from archaea, E. coli (Blattner et al. 1997), H. influenzae (Fleischmann et al. 1995), Helicobacter pylori(Tomb et al. 1997), B. subtilis (Kunst et al. 1997),Mycoplasma genitalium (Fraser et al. 1995), Mycoplasma pneumoniae (Himmelreich et al. 1996), Mycobacterium tuberculosis (Cole et al. 1998), Borrelia burgdorferi(Fraser et al. 1997), T. pallidum (Fraser et al. 1998),A. aeolicus (Deckert et al. 1998), and Synechocystissp. PCC6803 (Kaneko et al. 1996) from bacteria, and S. cerevisiae (Goffeau et al. 1997) from eukarya. The amino acid sequence data and the information of gene locations (ORFs) were taken from the complete genomes section of GenBank (ftp://ncbi.nlm.nih.gov/genbank/genomes/) as incorporated in the GENES database in KEGG (http://www.genome.ad.jp/kegg/). We accepted the authors' ORF assignments except for P. horikoshii; we removed 252 shadow genes which were entirely embedded in longer ORFs on the other strand (Kawarabayasi et al. 1998). The information of functional annotations is taken from KEGG (Ogata et al. 1999) and SWISS-PROT (Bairoch and Apweiler 1998), together with some new functional assignments we made using the sequence similarity search.
Identification of Membrane Proteins
Membrane proteins were identified from the sets of ORFs by means of two complementary automatic procedures followed by manual verification (Fig. 4). One is to use discriminant function to detect highly hydrophobic regions in the amino acid sequence, whereas the other is to rely on sequence similarity to known membrane proteins. In the first procedure, an amino-terminal signal peptide has to be properly removed because it is a hydrophobic segment often mislabelled as a transmembrane segment by any predictive method of membrane proteins. The prediction of signal peptides is based on a method similar to PSORT (Nakai and Kanehisa 1991; Nakai and Horton 1999), which consists of two steps: first to identify the existence of a signal peptide by amino acid sequence features (McGeoch 1985), and then to detect the cleavage site using a weight matrix (von Heijne 1986).
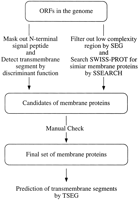
A schematic illustration of the procedures to identify membrane proteins in the complete genome.
After removal of the signal peptide, we employed discriminant analysis for distinguishing between a membrane protein and a
globular (nonmembrane) protein. The discrimination function was constructed for the most hydrophobic 17-residue region of
a protein sequence from the training sets of true (membrane proteins) and false (globular proteins) data (Kihara and Kanehisa 1997). The set of membrane proteins was extracted from the SWISS-PROT database release 34.0 (Bairoch and Apweiler 1998). When fragment entries were excluded and only one entry was selected from those with >30% of sequence identity, the data
set contained 3251 sequences. The set of globular proteins was based on the PDBSELECT database 97-March version (Hobohm et al. 1992) excluding entries of membrane and lipid associated proteins. The 35% threshold list was used and the data set contained
928 sequences. The resulting discrimination formula was as follows: where denotes the average hydrophobicity of the 17-residue region using the Kyte-Doolittle (1982) hydrophobicity index. An ORF is predicted to be a membrane protein if the function is positive. We chose the 17 residue-long
window size because it discriminated between the two training sets best, correctly assigning 94.3% of membrane proteins and
95.2% of globular proteins (detailed data not shown).
where denotes the average hydrophobicity of the 17-residue region using the Kyte-Doolittle (1982) hydrophobicity index. An ORF is predicted to be a membrane protein if the function is positive. We chose the 17 residue-long
window size because it discriminated between the two training sets best, correctly assigning 94.3% of membrane proteins and
95.2% of globular proteins (detailed data not shown).
The advantage of the first procedure is that it is an ab initio type prediction without relying on sequence similarity, but the prediction accuracy is not necessarily very high. To compensate the drawback of the first procedure, we also employed sequence similarity searches in the second procedure (Fig. 4). We used the SSEARCH program (Pearson 1991) against the SWISS-PROT database with the default parameter setting, after preprocessing of the query sequence using the SEG program (Wooton and Federhen 1993) with the default setting. SEG is effective to filter out low-complexity regions, which are stretches of hydrophobic amino acids in our case, and to reduce spurious hits (Bork and Koonin 1998). Each ORF is used as a query sequence, and it is considered as a possible membrane protein when the database hit includes any of the definite membrane proteins satisfying either of the following two criteria. The first criterion is that the E value does not exceed 0.001 (Brenner et al. 1995). In the other criterion, the E value may be ⩽0.1, but the Smith-Waterman score must be ⩾120, the Z-score must be ⩾120, and the alignment overlaps ⩾50% of the sequence lengths. The ORF sequences selected by the two procedures were then checked manually.
Prediction of Transmembrane Segments
We used the TSEG program (Kihara et al. 1998) for prediction of transmembrane segments. The basic idea of the method is to classify transmembrane segments into five types according to the average hydrophobicity and the periodicity of hydrophobicity and to model the membrane protein group with a specific number of transmembrane segments in terms of a series of different transmembrane segment types. For example, the models of membrane protein groups with one to nine transmembrane segments are, respectively, (1), (2,2), (2,3,3), (2,2,3,2), (2,3,3,2,2), (2,2,2,2,3,2), (2,3,3,2,2,2,5), (2,3,2,2,2,2,4,2), and (2,2,2,2,2,2,4,5,2) where 1–5 represent the types of transmembrane segments, 1 being most hydrophobic and 5 being least hydrophobic. The prediction of transmembrane segments involves selection of the most compatible model among different models, including one for globular proteins.
Identification of ATP-binding Proteins
The identification of ATP-binding proteins was based on the motif search. If an ORF sequence contains the P-loop ATP/GTP binding
motif of the PROSITE database (Hofmann et al. 1999): then it is considered as an ATP-binding protein.
then it is considered as an ATP-binding protein.
Identification of Conserved Clusters
SEG and SSEARCH were used to identify membrane proteins and ATP-binding proteins that correspond to each other in two clusters. Two clusters were considered to be conserved if >1 pair of constituent proteins is similar. The Smith-Waterman score of 150 was used for the similarity criterion. Generally, the threshold score of 120 is high enough to detect related sequences (Pearson 1996, 1998). However, according to our experience, obviously unrelated sequences were found for both membrane and ATP-binding protein searches when the threshold score of 120 was used. Conserved clusters were collected into a group by single-linkage clustering; namely, a cluster was added to the group if it was similar to at least one of the clusters in the group.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research on the Priority Area Genome Science from the Ministry of Education, Science, Sports, and Culture of Japan. The computational resource was provided by the Supercomputer Laboratory, Institute for Chemical Research, Kyoto University.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








