
Evaluation of Single Nucleotide Polymorphism Typing with Invader on PCR Amplicons and Its Automation
- Charles A. Mein1,
- Bryan J. Barratt1,
- Michael G. Dunn1,
- Thorsten Siegmund1,
- Annabel N. Smith1,
- Laura Esposito1,
- Sarah Nutland1,
- Helen E. Stevens1,
- Amanda J. Wilson1,
- Michael S. Phillips2,
- Nancy Jarvis3,
- Scott Law3,
- Monika de Arruda3 and
- John A. Todd1
- 1Wellcome Trust Centre for the Study of Molecular Mechanisms in Disease, University of Cambridge, Addenbrooke's Hospital, Cambridge CB2 2XY UK; 2Third Wave Technologies, Inc., Madison, Wisconsin 53719 USA; 3Merck Research Laboratories, West Point, Pennsylvania 19486 USA
Abstract
Large-scale pharmacogenetics and complex disease association studies will require typing of thousands of single-nucleotide polymorphisms (SNPs) in thousands of individuals. Such projects would benefit from a genotyping system with accuracy >99% and a failure rate <5% on a simple, reliable, and flexible platform. However, such a system is not yet available for routine laboratory use. We have evaluated a modification of the previously reported Invader SNP-typing chemistry for use in a genotyping laboratory and tested its automation. The Invader technology uses a Flap Endonuclease for allele discrimination and a universal fluorescence resonance energy transfer (FRET) reporter system. Three hundred and eighty-four individuals were genotyped across a panel of 36 SNPs and one insertion/deletion polymorphism with Invader assays using PCR product as template, a total of 14,208 genotypes. An average failure rate of 2.3% was recorded, mostly associated with PCR failure, and the typing was 99.2% accurate when compared with genotypes generated with established techniques. An average signal-to-noise ratio (9:1) was obtained. The high degree of discrimination for single base changes, coupled with homogeneous format, has allowed us to deploy liquid handling robots in a 384-well microtitre plate format and an automated end-point capture of fluorescent signal. Simple semiautomated data interpretation allows the generation of ∼25,000 genotypes per person per week, which is 10-fold greater than gel-based SNP typing and microsatellite typing in our laboratory. Savings on labor costs are considerable. We conclude that Invader chemistry using PCR products as template represents a useful technology for typing large numbers of SNPs rapidly and efficiently.
Single-nucleotide polymorphisms (SNPs) are the most common form of genetic polymorphism. This, coupled with their potential as functional variants, has produced a great deal of interest in SNPs both as pharmacogenetic indicators and as markers for mapping genes for complex diseases (Risch and Merilangas 1996; Kruglyak 1997;Masood 1999). A large number of SNPs have already been identified with >21,000 entries on the NCBI's SNP database alone (http://www3.ncbi.nlm.nih.gov/SNP/). Many recent studies are focused on identifying polymorphisms that lie in the coding sequence of potential candidate genes for common diseases (Nickerson et al. 1998;Camien et al. 1999; Cargill et al. 1999; Halushka et al. 1999). The ability to genotype this abundant source of variation rapidly and accurately is becoming an ever more important goal in the genetics community (Bonn 1999). A variety of technologies available have the potential to transfer to high-throughput genotyping laboratories (Landegren et al. 1998). These include 5′ exonuclease assays, such as TaqMan (Livak et al. 1995), molecular beacons (Tyagi et al. 1998), Oligonucleotide-ligation assays (OLAs) (Tobe et al. 1996), dye-labeled oligonucleotide ligation (DOL) (Chen et al. 1998), minisequencing (Chen and Kwok 1997; Pastinen et al. 1997), microarray technology (Hacia et al. 1998; Wang et al. 1998), mass spectroscopy (Ross et al. 1998) and the scorpions assay (Whitcombe et al. 1999). However, no single chemistry has gained acceptance as the technology of choice. A suitable method for such applications must be accurate and homogenous, develop a robust, easily interpretable signal, and be flexible enough to extend to novel loci with little optimization. These features will lend the technology to automation.
The invasive cleavage of probe oligonucleotides has been used to genotype SNPs using genomic DNA as template (Lyamichev et al. 1999;Ryan et al. 1999). Conscious of the absolute need to conserve stocks of genomic DNA template, we have modified the technique to use PCR products as template (PCR-Invader assay). The Invader technology relies on the specificity of recognition and cleavage by a Flap Endonuclease (FEN) of the three-dimensional structure formed when two overlapping oligonucleotides, an Invader oligonucleotide, and a signal oligonucleotide with a reporter arm hybridize to target DNA containing a polymorphic site (Lyamichev et al. 1999). Only in the presence of a perfect match between signal probe and template is the signal probe reporter arm, or flap, cleaved to drive a universal secondary cleavage reaction with a fluorescence resonance energy transfer (FRET) label (Ryan et al. 1999). Signal is detected at an end point with a conventional fluorescence microtitre plate reader.
To test the robustness and accuracy of the PCR-Invader assay in a high-throughput environment, assays were developed for a panel of 36 SNPs and one 5-bp deletion/insertion polymorphism. The polymorphisms were drawn from published loci that are either candidate genes for, or associated with, type 1 diabetes susceptibility or unpublished polymorphisms from LRP5 (Hey et al. 1998; R. Twells, M. Phillips, and J.A. Todd, unpubl.) and ESTs on chromosome 11q13 (Methods; Table 1). Markers were typed in 384 individuals using both PCR-Invader and conventional assays (restriction fragment length polymorphism, RFLP), forced RFLP (cRFLP) (Li and Hood 1995), amplification refractory mutation PCR (ARM–PCR), and fluorescently labeled length polymorphism) to measure genotyping accuracy. Individuals that gave inconsistent genotypes between PCR-Invader technology and the conventional method were reassayed with both methods to confirm the genotype.
Invader Assay Design Summary
RESULTS
Assays were successfully designed for all 37 polymorphisms; of these 30 (81%) passed the initial design quality control criteria at the first attempt (Table 1). Six of the outstanding loci were successful at the second attempt either by switching target strand or by changing the reporter arm (Table 1). The remaining assay,EST4c3021–3026del, required a third design attempt (Table 1). Designs 2 and 3 differ by an additional 3 bases at the 5′ end of the Invader oligonucleotide (Table2).
Loci-Specific Oligonucleotide Sequences for the Invader Assay
Initially, PCR-Invader reactions were set up in a 96-well format. The results of a typical locus, CTLA4 g.49A>G, for 96 individuals are shown in Figure 1. A plot of gross signal from the allele A against that from allele G shows four distinct clusters of points reflecting the three possible genotypes (homozygous A, homozygous G, and heterozygotes) and two PCR failures toward the origin (Fig. 1a). The same data are displayed as a ratio of A to G signal (Fig. 1b). Figure 1 clearly demonstrates the level of discrimination between genotypes with ratios of 9.86 ± 3.10 and 0.07 ± 0.04 for A and Ghomozygotes, respectively, and 1.13 ± 0.34 for heterozygotes. Average ratios for other loci tested are shown in Figure2. For all but one locus, there is a clear distinction between the three different genotypes. Only CTLA4 g.-318C>T has overlapping ranges between the heterozygote population and individuals homozygous for the T allele. However, there are only three individuals homozygous for the Tallele in the test population and one of those had an anomalous ratio, 0.54 compared with 0.13 and 0.17. The average ratio of signal between each allele across all loci is 9.04 ± 3.5 for homozygotes and 1.1 ± 0.34 for heterozygotes. There is variation in ratios between loci. For example, the heterozygote ratio for EST3 c.448G>Aof 2.25 ± 0.49 differs considerably from the average ratio of 1.1 ± 0.34. This variation may be produced by a number of factors, including differential annealing temperatures of the two-allele-specific probes, varying efficiencies of cleavage, or preferential amplification of one allele during PCR. The interlocus difference in ratios makes it impossible to produce all locus criteria for scoring SNP genotypes. However, each locus tested had a characteristic pattern of ratios reproducible between runs and can be used to generate empirical criteria for assigning genotype (Fig. 2).
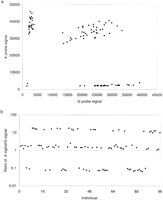
Data from 96 individuals for the marker CTLA4 g.49A>Gplotted as gross signal from each allele (a) or as a ratio of A signal to G signal (b). Data were acquired from the 96-well format PCR-Invader assay.
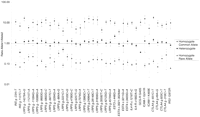
Average ratios, ±1 s.d., from 384 individuals, of signal from common allele to rare allele for each genotype for each locus. Data were generated in a 96-well PCR-Invader format.
Failure to score the genotype of an individual can lead to time-consuming and costly repeat experiments or loss of informativity for mapping experiments. The failure rate for each locus ranges from 0.5% to 7.3%, average 2.3% (Table 3). The failures seen were due to samples not amplifying at the PCR stage of the protocol.
Summary of Loci and Number of Discordant Typings
To test the accuracy of the PCR Invader assay we compared typings with those from other commonly used typing technologies. Conventional genotyping assays (RFLP, cRFLP, ARMs, and fluorescently labeled length polymorphism) were developed for 31 of the polymorphisms. Ninety-nine percent (99.18%; 11,806 of 11,904 total genotypes) of genotypes were concordant between the conventional assay and PCR-Invader assays (Table3). Individuals with discordant typings were retyped with both technologies to confirm genotypes. Each of the individuals with discordant genotypes was typed a minimum of four times, twice with each technology. In all but one case, a consensus genotype was established from which it was possible to elucidate the reason for the original anomaly. Forty-five of the discrepancies (46% of the total) could be attributed to incomplete digestion in the PCR–RFLP assay or allele failure in the ARMs-PCR assay (Table 3). Forty-nine mistypings (50%) of discordant typings were found to be false homozygotes produced during the PCR-Invader assay (Table 3). It is most likely that these reflect a failure to deliver template to one allele of the assay. Four cases were seen, each from different loci, of PCR-Invader producing false-positive results, that is, a homozygote typed as a heterozygote (Table 3). This represents 4% of the total error rate and may reflect PCR contamination in the initial Invader reaction. A single individual consistently typed differently between the PCR-Invader assay and the PCR–RFLP at locus CTLA4 g.49A>G. The reason for this is unclear but may represent a previously unknown polymorphism adjacent to the assayed polymorphism that affected hybridization of one of the Invader probes.
We failed to develop reliable conventional assays for the remaining six loci. All of these loci are located within the 200-kb genomic sequence flanking and including LRP5, and are adjacent to other loci in this study or to microsatellites and SNPs typed previously in our laboratory (R. Twells, C.A. Mein, and J.A. Todd, unpubl.). However, evidence of linkage disequilibrium (LD) between these and adjacent markers was detected (data not shown), suggesting that these loci were typed accurately.
A single locus, LRP5 g.35592T>C, proved difficult to amplify and gave a smear when PCR products were visualized by Agarose gel electrophoresis (data not shown). Despite this result, the PCR-Invader reaction produced easily interpretable genotypes (Fig. 2).
A potential problem for any DNA typing technology is the presence of sequences elsewhere in the genome that share significant sequence similarity with the target sequences, such as repetitive elements, pseudogenes, and other gene family members. In our test panel, 11 of the loci selected from the LRP5 region of chromosome 11q13 share sequence similarity of >58% with the BAC clone, bK246H3, from chromosome 22q11.21-q12 (accession no. AL022324), which contains a pseudogene of LRP5 (Table 3). There are also seven loci that show a significant amount of sequence similarity with the consensus sequence of various Alu repeat subfamilies detected by RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker). Despite this sequence similarity to other genomic segments, all 12 SNP loci gave reliable and consistent genotypes (Fig. 2; Table 3). Notably, there was no difference in the signal-to-noise ratios between those loci with similarity to other genome sequences and those that appear to be unique sequences (Fig. 2; Table 3).
This degree of fidelity in PCR-Invader assays is produced by the requirement for multiple oligonucleotide hybridizations in both PCR and Invader reactions and the exquisite specificity of FEN.
Genotyping of loci in regions that have a low GC content may present difficulties for some technologies (Chen et al. 1998). The test set of loci we have used in this study ranges in GC content from 24% to 68% (Table 3). There was no difference in the efficiency of genotyping as measured by the number of mistypings between conventional assays and PCR-Invader assay or as measured by the relative signal intensities for the three genotypic states (Fig. 2; Table 3).
Some of the loci under investigation were <1 kb distant from each other. PCR primers were designed so that such adjacent loci were contained in the same amplicon. In total, 12 such loci were contained in five fragments (Table 4). Each locus within a PCR fragment gave genotypes of similar quality (Fig. 2; Table 3). Therefore, the effective amount of genomic DNA used as template per assay can be reduced, perhaps to below 1 ng, by utilizing a combination of multiplex and long-range PCR. An alternative strategy to reduce initial template is to use PCR products generated from template derived from degenerate PCR amplification (Dunger et al. 1998). Preliminary results for PCR-Invader assays from such templates indicate success for some loci but more variability than seen with PCR from genomic DNA, indicating problems with PCR fidelity.
PCR Primer Sequences for Invader and Conventional Assays
The successful results with the PCR-Invader assay in a 96-well format encouraged us to transfer the reactions to a 384-well format on a semiautomated platform. To evaluate the fidelity of an automated 384-well-based system, we typed a subset of nine loci. All nine assays tested in a 384-well format gave fully interpretable genotypes (Fig.3). The average signal ratio between each allele across all nine loci is 7.53 ± 0.72 for homozygotes and 1.01 ± 0.07 for heterozygotes. This compares with values of 9.00 ± 1.53 and 1.01 ± 0.18 for the same nine loci when assayed in the 96-well format. Although the signal-to-noise ratio for homozygous individuals was slightly less in the 384-well format than in the 96-well format, the coefficient of variance (CV) was lower, 9.55% and 16.96%, respectively. This reduced variability will lead to increased confidence in scoring genotypes. Both formats showed a similar rate of assay failure, 3.03% in the 384-well format and 2.78% for the same loci in 96-well format (Table 3). Four individuals (0.14%) scored differently in a 384-well assay compared with genotypes from 96-well Invader assays or conventional typings (Table 3). Three of these cases were known heterozygotes scored as homozygotes. The fourth individual was a known homozygote typed as a heterozygote. The explanation for these variant results is probably identical between the 96- and 384-well formats, namely, a failure to deliver template to some wells and a low level of contamination between samples.

Average ratios, ±1 s.d., from 384 individuals, of signal from common allele to rare allele for each genotype for 9 loci in 384- and 96-well formats.
DISCUSSION
The results of this study clearly indicate that the PCR-Invader assay is a useful tool for high-throughput genotyping. Successful PCR-Invader assays were developed for all of the polymorphisms in our test panel. This success compares favorably with our attempts to conventionally type the same polymorphisms with 6 of 36 (17%) remaining untyped. The accuracy of the panel of PCR-Invader assays used, 99.2%, is good; the accuracy of other SNP typing technologies is largely unknown. The failure rate of the PCR-Invader assay is relatively low, 2%–3%, and probably reflects the underlying rate of PCR failure. The failure rate is similar to that observed for DOL (Chen et al. 1998), although the failure rate for other technologies is not known. Preliminary experiments with TaqMan assays suggested between 5% and 15% failure/repeat rate depending on the SNP assay (A.J. Wilson and J.A. Todd, unpubl.).
PCR-Invader technology has several features that suit its use for the high-throughput genotyping environment. The assay is performed entirely in a microtitre format with a single addition of Invader reagents to PCR products, generating a stable fluorescent signal that can be captured at an end point followed by a simple data interpretation step. No gel electophoresis, purification, or manual data entry is required, thereby reducing the error rate. Most of the assays (81%) were successfully designed at the first attempt, and no assay required more than two redesigns. This contrasts with other assays that may prove difficult to expand to large numbers of loci (Chen et al. 1998). Optimization of each new assay is straightforward with a temperature titration for both PCR and Invader assay steps, although this would become disadvantageous with larger numbers of assays. Advances in algorithms to predict the melting temperature used in the design of the Invader assay now allow all Invader reactions to be run at the same temperature (Third Wave Technologies, Inc., H. Allawi, in prep.).
The synthesis of fluorescently labeled probes specific for each locus, as in the TaqMan assay (Livak et al. 1995) can be prohibitively expensive. In the current study, only two fluorescently labeled oligonucleotides were used for the 37 assays, significantly reducing costs. Further developments from this work have allowed the use of a single-arm sequence with all SNP loci (Third Wave Technologies, Inc., unpubl.). This allows a universal reporter system to be dried down to the surface of microtitre plates ready for the addition of sequence-specific oligonucleotides and template.
However, the most significant advantage of this technology is its flexibility and robustness. For pharmacogenetic testing and in fine mapping projects, it is important to have assays available irrespective of the position of the base of interest in the genome or sequence composition. Many etiological polymorphisms are likely to lie at the 5′ end of genes that are known to have a high GC content or lie in regions with similarity to sequence elsewhere, for example, conserved sequence motifs, binding motifs, or pseudogenes. Unusual base compositions and similarities to other regions are likely to confound technologies that rely purely on hybridization for specificity. PCR-Invader assays were designed successfully for all loci tested, including those with distorted GC content and 14 loci with similarity to other genomic segments.
One disadvantage with the current technology is the need to assay each allele separately. Ninety-two percent of the initial mistypings with the PCR-Invader assay are assumed to be a failure to deliver template to one of the reactions, leading to heterozygous individuals being mistyped as homozygotes. A lack of an internal control is, of course, not a flaw unique to the PCR-Invader assay. For example, it is not always possible to design an RFLP assay with a control restriction site leading to the same bias. Addition of an inert dye, such as cresol red, to the PCR will allow a visual check on template delivery to the Invader reaction. Alternatively, the development of more fluorescent dyes will allow the detection of both alleles in the same reaction and each acting as a control for the other would eliminate this problem and could also allow multiplexing of assays.
SNP-based genetic maps used for gene mapping projects will need many more markers than the current microsatellite maps due to the lower heterozygosity of SNPs. A genetic map of 700–900 SNPs is required to give equivalent information to the maps on the basis of 300–400 microsatellite loci used currently (Kruglyak 1997). A genome scan of 400 sib pair families will therefore require the generation of 1–1.5 million genotypes. The ability to automate SNP typing will make such projects feasible. We have successfully installed a semiautomated SNP typing platform on the basis of two liquid handling robots and a fluoresence microtiter plate reader with automated loading. This system, with capital costs similar to a large microsatellite genotyping project, can generate at least 50,000 genotypes in a 5-day period with two to three operators. Hence, it will be possible to perform a genome scan of the size mentioned in under 4 months, a project we estimate would take 12 months with microsatellite markers.
METHODS
DNA Samples
A total of 384 samples were drawn from 96 type 1 diabetic families, part of the British Diabetic Association-Warren repository (Bain et al. 1990). DNA was extracted from Epstein–Barr virus (EBV)-transformed peripheral blood lymphocytes. Briefly, 50 ml of confluent EBV cells was pelleted at 1000 rpm for 5 min. The supernatant was discarded and cells were resuspended in 4 ml of 5.25 m guanidine hydrochloride (Sigma), 0.5 m ammonium acetate (Sigma), 0.5 mg of proteinase K (Sigma), and 0.3% sodium sarcosyl (Sigma). The solution was incubated overnight at 37°C. Two milliliters of chloroform was added and spun to 2500 rpm, the upper layer was removed and added to 10 ml of 100% ethanol, and the precipitated DNA was pelleted at 3500 rpm in an Allegra 6R microcentrifuge (Beckman, UK). Pellets were washed with 70% ethanol and resuspended in 300 μl of Tris-EDTA (TE) (pH 7.5). DNA was quantated with Pico Green (Molecular Probes, Eugene, OR) and diluted to 4 ng/μl in TE (pH 7.5) before use.
Panel of SNPs
Twenty six of the SNPs selected are a subset of 79 novel polymorphisms generated by comparing genomic sequence information from two unrelated individuals from a 300-kb region on chromosome 11q13 (R. Twells, M. Phillips, and J.A. Todd, unpubl.). Fifteen of these represent all of the polymorphisms detected in a 21-kb stretch around the first exon of LRP5 that had not been typed by us previously. Three SNPs and the 5-bp-length polymorphism are derived from two ESTs in the region, and the remaining were selected at random. Other polymorphisms were selected from the literature: ICAM1(Vora et al. 1994); IRS1 (Almind et al. 1993); GCGR(Hager et al. 1995); CTLA4 (Deichmann et al. 1996;Nisticó et al. 1996); IL4r (Deichmann et al. 1997;Hershey et al. 1997); and INS (Ullrich et al. 1980) as potential, or proven in the case of INS (Bennett et al. 1995), susceptibility loci for type 1 diabetes. All of the loci were selected without reference to the surrounding sequence composition.
PCR
PCR primers were designed, using the program Primer 3 (Rozen and Skaletsky 1997), to amplify at least 50 bp on either side of the polymorphic site. Five larger PCR fragments were designed to include multiple polymorphic sites (a; LRP5 g.-11215T>A,LRP5 g.-11094G>A, b; LRP5 g.-10088G>A, LRP5 g.-9693GA, c;LRP5 g.-5802G>C, LRP5 g.-5677C>T, LRP5 g.-5264C>A, d;EST4 c.3021-3026del, EST4 c.3211G>A,EST4 c.3403G>A, and e IL4-Rc.1216T>C andIL4-Rc.1902A>G). The average amplicon length, excluding fragments containing multiple sites, was 397 bp (Table 4). PCR conditions were optimized by varying MgCl2 concentrations between 1 and 5 mm and annealing temperature between 45°C and 65°C. Ninety-six-well microtiter plate PCRs were performed in thin-walled polycarbonate microtiter plates (Corning Costar, Corning, NY, US). Two and one-half microliters of 4 ng/μl stock of genomic DNA was dispensed into each well with a 96-syringe Hydra (Robbins, Sunnyvale, CA). Five microliters of PCR reaction mix containing 0.2 mm dNTP, 2 ng/μl forward and reverse primer, and 0.375 units of TaqGold (Perkin Elmer Applied Biosystems, Foster City, CA), was added and the reaction overlaid with 10 ml of mineral oil (Sigma). Reactions were incubated at 94°C for 14 min, and 35 cycles of 94°C for 30 sec, annealing temperature (dependent on locus) for 30 sec, and 72°C for 30 sec on MJ PTC225 thermocyclers (MJ Research, Watertown, MA). The 384-well reactions were performed in polypropylene plates (Advanced Biotechnologies) in 6-μl final reaction volumes using the same conditions as the 96-well format. Plates were heat sealed with easy-peal strong foil seals (Advanced Biotechnologies) with PCR cycling conditions as before. All pipeting steps for PCR preparation were performed with a Hamilton 2200 liquid handler dedicated to pre-PCR work.
Invader Assay
Probe sets for each locus were designed and synthesized by Third Wave Technologies, Inc. (Madison, WI) (Lyamichev et al. 1999; Ryan et al. 1999) (Tables 2 and 5). Target-strand selection was based on avoiding polynucleotide tracts in the primary probe. The Invader oligonucleotide and primary probe were designed to have theoretical annealing temperatures of 80°C and 65°C, respectively, using a nearest neighbor algorithm on the basis of final probe and target concentrations. The Invader oligonucleotides were designed so that the 3′ base overlaps with the target polymorphism, but is not complementary to either allele. The Primary Probes were designed with one of four possible reporter arms selected to avoid secondary structure between arm sequence and that of the target template-specific region of the probe. A 3′ amine was added to each Primary Probe to prevent uncleaved signal oligonucleotide acting as an Invader oligonucleotide in the secondary reaction. Secondary reactions composed of a fluorescein and cy3-labeled FRET probe and a secondary target oligonucleotide were designed with an optimal annealing temperature of 55°C (Ryan et al. 1999; Table 5). In total, two FRET probes and five secondary oligonucleotides were used, the choice of which was based on the sequence of the locus tested (Table 5). All oligonucleotides with the excpetion of Invader oligonucleotides were purified by ion-exchange chromatography. Empirical optimal annealing temperatures for each of the specific probe sets were determined by performing the reaction on synthetic target oligonucleotides at six separate temperatures between 60°C and 75°C. Signal strength of at least 80% maximum was seen 2°C either side of the maximum for all loci tested. Loci were initially considered to have passed the design stage if the signal from the synthetic target was fourfold greater than that from the no target. Subsequently, these criteria have been modified so that an assay is considered to have passed the initial quality control if the signal from the synthetic target is >1.5 times greater than the standard deviation of the signal from the no target control. Seven assays did not pass the initial design criteria. These loci were redesigned either to the opposite strand or with an alternative reporter arm (Tables 1 and 2;http://www.gene.cimr.cam.ac.uk/todd/HumData/mein_et_al_2000).
Oligonucleotide Sequences for the Secondary Invader Reaction
Assays were prepared for each allele separately. In a 96-well format, PCR products were diluted 1 in 200 or 1 in 20, determined empirically dependent on locus, in RNase-free water using a 96-syringe Hydra (Robbins, Sunnyvale, CA). Two 3-μl aliquots were dispensed with a 96-syringe Hydra (Robbins) into 96-well thin-walled polycarbonate microtiter plates (Corning Costar). PCR product was dried for 15 min at 80°C. Five microliters of Invader mix, consisting of 4% PEG-8000, 10 mm MOPS, 0.025 μm Invader oligonucleotide, 0.25 μm secondary target oligonucleotide, 0.5 μm FRET probe, 5 ng/μl Escherichia coligenomic DNA as a carrier, 0.05 μm Primary Probe 7.5 mm MgCl2 and 2.5 ng/μl FEN (all reagents from Third Wave Technologies) were added to the dried PCR products. The samples were overlaid with 5 μl of mineral oil (Sigma) and incubated at 95°C for 5 min, 63°C–71°C (dependent on locus; Table 1) for 10 min and 55°C for 10 min.
In the 384-well format, PCR products were diluted 1 in 4 in RNase-free water. Two 2-μl aliquots were made into 384-polycarbonate PCR plates (Advanced Biotechnologies) and 4 μl of each Invader mix was added, consisting of 4% PEG-8000, 10 mm MOPS, 0.025 μm Invader oligonucleotide, 0.25 μmsecondary target oligonucleotide, 0.5 μm FRET probe, 5 ng/μl E. coli genomic DNA as a carrier, 0.05 μm Primary Probe 7.5 mm MgCl2 and 0.5 ng/μl FEN (all reagents purchased from Third Wave Technologies). Plates were heat sealed with easy peal strong foil (Advanced Biotechnologies) and incubated at 95°C for 5 min, 63°C–71°C (dependent on locus) for 10 min, and 55°C for 10 min on MJ PTC225 thermocyclers (MJ Research). All pipeting was performed with a Multimek liquid handling robot (Beckman, Coulter, Allendale, NJ) fitted with 50-μl disposable tips and an automatic wash station.
Fluorescence was measured directly at the end of incubation using a Cytofluor 4400 fluorescence microtiter plate reader (Perkin Elmer), excitation 485/20, emission 530/25, and gain 60. Results were analyzed using Excel software (Microsoft, Redmond, WA). Individual genotypes were scored by taking a ratio of signal strength from allele 1and allele 2. The criteria for scoring genotypes varied between loci (see Results), but for most loci, an individual was typed as a heterozygote if the ratio of signal between the two alleles was between 0.5 and 2. Ratios outside of this range were typed as homozygotes. An assay was classed as a failure if signals from both assays were below a threshold level dependent on locus or format.
RFLP Assays
cRFLPs for loci with no suitable RFLP assay were designed with mismatched primers to generated restriction sites (Cohen and Levinson 1988). Restriction enzymes were obtained from New England Biolabs. Five microliters of PCR product was digested overnight with 0.5 units of enzyme per reaction in a total volume of 10 μl, under the manufacturers conditions. Digestion products were separated on 2.5%–5% Agarose (FMC BioProducts, Rockland, ME), depending on fragment size, and visualized by staining with ethidium bromide.
Length Polymorphism
EST4 c.3021–3026del was amplified with a 5-FAM-labeled forward primer under standard PCR conditions. PCR product was diluted 1 in 10 and 1 μl added to 1 μl of gel-loading buffer [5:1 formamide to 50 mm EDTA (pH 8), containing 50 mg/ml blue dextran]. One microliter was loaded onto an ABI 377 automated sequencing gel (6% acrylamide 19:1 acrylamide: bis acylamide) (FMC BioProducts) and analyzed using Genescan and Genotyper packages (Perkin Elmer Applied Biosystems).
Evaluation of Genotyping Accuracy
Individuals that gave discordant genotypes between the Invader technology and the conventional typing methodology along with the other members of the pedigree were retyped using both techniques.
Acknowledgments
This work was funded by the Wellcome Trust, Juvenile Diabetes Foundation International, the British Diabetes Associations, and Merck Research Laboratories. B.J. Barratt is a UK Medical Research Council CASE student, partly funded by Oxagen. We thank R. Twells forLRP5 polymorphism information and for discussion.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL john.todd{at}cimr.cam.ac.uk; FAX 44-2–334762-102.
-
- Received November 8, 1999.
- Accepted January 18, 2000.








