
Functional Classification of cNMP-binding Proteins and Nucleotide Cyclases with Implications for Novel Regulatory Pathways in Mycobacterium tuberculosis
Abstract
We have analyzed the cyclic nucleotide (cNMP)-binding protein and nucleotide cyclase superfamilies using Bayesian computational methods of protein family identification and classification. In addition to the known cNMP-binding proteins (cNMP-dependent kinases, cNMP-gated channels, cAMP-guanine nucleotide exchange factors, and bacterial cAMP-dependent transcription factors), new functional groups of cNMP-binding proteins were identified, including putative ABC-transporter subunits, translocases, and esterases. Classification of the nucleotide cyclases revealed subtle differences in sequence conservation of the active site that distinguish the five classes of cyclases: the multicellular eukaryotic adenylyl cyclases, the eukaryotic receptor-type guanylyl cyclases, the eukaryotic soluble guanylyl cyclases, the unicellular eukaryotic and prokaryotic adenylyl cyclases, and the putative prokaryotic guanylyl cyclases. Phylogenetic distribution of the cNMP-binding proteins and cyclases was analyzed, with particular attention to the 22 complete archaeal and eubacterial genome sequences. Mycobacterium tuberculosis H37Rv andSynechocystis PCC6803 were each found to encode several more putative cNMP-binding proteins than other prokaryotes; many of these proteins are of unknown function. M. tuberculosis also encodes several more putative nucleotide cyclases than other prokaryotic species.
Signal transduction pathways control many critical cellular processes, including chemotaxis, differentiation, proliferation, and apoptosis. For example, signal transduction pathways are necessary for bacterial pathogens to sense and respond to host environments, cellular differentiation during embryogenesis, conductance of nerve impulses, and cell cycle control. Disruption of these pathways can result in neoplasia, arteriosclerosis, neurological and developmental abnormalities, and cell death. The most common mechanisms of signal transduction include the phosphorylation or dephosphorylation of effector proteins by kinases and phosphatases, respectively, and the production of second messengers. Cyclic nucleotides were first recognized as second messengers 40 years ago. Such diverse molecules as (p)ppGpp, Ca2+, inositol triphosphate, and diacylglycerol have also been recognized as second messengers since then.
The cyclic nucleotides adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP) are key universal second messengers, mediating cellular functions in organisms as phylogenetically diverse as Escherichia coli and Homo sapiens. Intracellular concentrations of cyclic nucleotides (cNMPs) are controlled by regulation of their relative rates of synthesis, excretion, and degradation (Botsford and Harman 1992; Tang et al. 1998). The nucleotide cyclases (adenylyl and guanylyl cyclase), the cNMP phosphodiesterases, and the cyclic nucleotide effector proteins (cNMP-binding proteins) have been particularly intense areas of signal transduction research, providing detailed studies of these proteins (for reviews, see Kolb et al. 1993; Bârzu and Danchin 1994;Francis and Corbin 1994; Beavo 1995; Finn et al. 1996; Tang et al. 1998). The molecular mechanisms of cNMP export, however, are currently unknown.
cNMP-Binding Proteins
The cyclic nucleotide-binding proteins identified in prokaryotes consist of a small group of orthologous cAMPreceptor proteins (CRP) present only in gram-negative bacteria of the gamma subdivision of the Proteobacteria (Botsford and Harman 1992). CRP is a global regulator belonging to the CRP/FNR (Fumarate and NitrateReduction) family of prokaryotic transcription regulators. The CRP–cAMP complex is involved in positive as well as negative regulation of a wide variety of promoters (Botsford and Harman 1992;Kolb et al. 1993).
Three functional classes of cyclic nucleotide-binding proteins have been described in eukaryotes: kinases, channels, and guanine nucleotide exchange factors (GEFs). Cyclic nucleotide-dependent kinases have long been considered the primary effectors that mediate cellular responses to changes in intracellular cNMP concentrations (for review, seeFrancis and Corbin 1994). Both cAMP-dependent kinases (cAK) and cGMP-dependent kinases (cGK) have been described in many eukaryotic species. A significant number of cyclic nucleotide-gated and cyclic nucleotide-modulated ion channels are also involved in many cell functions in eukaryotes (for review, see Finn et al. 1996). In addition, mammalian cAMP-regulated guanine nucleotide exchange factors (cAMP–GEFs) that selectively activate Rap1A (a Ras family G protein) were recently described (Kawasaki et al. 1998).
All of these cNMP-binding proteins (cAK and cGK, cNMP-regulated channels, cAMP–GEFs, and CRP) share sequence homology in their cyclic nucleotide-binding domains, suggesting that they share structural similarity (Shabb and Corbin 1992). The crystal structures reported forE. coli CRP (Weber and Steitz 1987) and bovine cAK regulatory subunit (Su et al. 1995) support this hypothesis. The cNMP-binding domain of each of these proteins consists primarily of three α helices and an eight-stranded, antiparallel β-barrel. Proteins that bind cyclic nucleotides, but that are apparently unrelated to the proteins described above, have also been described (Hughes et al. 1988;Charbonneau et al. 1990; Grant and Tsang 1990; Firtel 1996). Whether any of these proteins share structural similarity to the cNMP-binding domain of CRP and cAK awaits structural studies.
Nucleotide Cyclases
Bârzu and Danchin (1994) described three classes of nucleotide cyclases, and Sismeiro et al. (1998) have recently described a fourth class. The largest and most diverse class, known as class III cyclases (for review, see Tang and Hurley 1998; Tang et al. 1998), includes all of the known cyclases from eukaryotes—the nine identified isoforms of mammalian adenylyl cyclase, the mammalian receptor and soluble guanylyl cyclases, the many cyclases identified in lower eukaryotes and fungi—as well as a number of prokaryotic cyclases. All class III cyclases exhibit significant homology in the catalytic region and are predicted to function as dimers of catalytic domains. The available crystal structures (Tesmer et al. 1997; Zhang et al. 1997) indicate that two cyclase catalytic domains interact in a head-to-tail fashion forming a wreathlike structure. These structures and mutational data have identified four amino acid residues that may be involved in catalysis and two residues that confer nucleotide specificity (Tang and Hurley 1998). Previous classifications of the class III cyclases have focused on their subcellular localization, protein topology, and means of activation (Tang and Hurley 1998; Tang et al. 1998).
We chose to examine the cNMP-binding proteins and nucleotide cyclases using recently developed Bayesian algorithms for multiple sequence alignment and database searching (PROBE; Neuwald et al. 1997; Liu et al. 1999), and classification (Classifier; Qu et al. 1998). These methods allowed us to identify many new members and present a novel classification for these protein superfamilies. We were able to use the volume of experimental data available for these proteins, in particular, X-ray crystal structures and mutational data, to compare structure–function data with our sequence analysis. Classification of the cNMP-binding protein family identified the known functional classes of this family, as well as indicating the existence of additional, previously unreported functional classes. Classification of the nucleotide cyclases identified two distinct prokaryotic classes, which may represent adenylyl and guanylyl cyclases for these species. Examination of these protein families with respect to the several complete genomes now available (22 prokaryotes, Saccharomyces cerevisiae, and Caenorhabditis elegans) revealed an interesting phylogenetic distribution with implications for horizontal transfer of genes.
RESULTS
cNMP-Binding Proteins
The seed sequence used with PROBE to identify the cNMP-binding protein superfamily was Streptomyces griseus P3 (gi‖1196910), a sporulation-specific, putative cNMP-binding protein (J. Kwak, L.A. McCue, K. Trczianka, and K.E. Kendrick, in prep.). After partial and duplicate sequences were removed, 207 sequences were left in the superfamily sequence set. The superfamily model consisted of three motifs, shown as sequence logos in Figure1. Included in the model were five strongly conserved glycines (Fig. 1: motif 1, positions 16, 23, and 35; and motif 2, positions 12 and 16) believed to be important for integrity of the β-barrel structure that forms the cNMP binding pocket (Weber and Steitz 1987; Su et al. 1995). Also included in the model were the glutamate (Fig. 1: motif 2, position 17) and arginine residues (Fig. 1: motif 3, position 2) that interact with cNMP (Weber and Steitz 1987; Su et al. 1995).
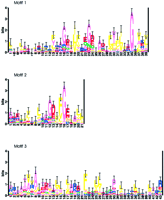
Sequence logos of the cNMP-binding protein superfamily. Sequences were aligned using PROBE with a purge value of 150. The average information at conserved positions is 0.915 bits. The conserved glycines important for β-barrel formation are located at: motif 1, positions 16, 23, and 35; motif 2, positions 12 and 16. cAMP contact residues are located at: motif 2, position 17; motif 3, position 2.
This superfamily consisted of many known cNMP-binding proteins, including eukaryotic cAKs and cGKs, cNMP-gated and cNMP-modulated channels, prokaryotic CRP proteins, and a putative cAMP–GEF. Also included in this superfamily were several prokaryotic transcription regulatory proteins (e.g., FNR, nitrogen fixation regulatory (FIXK), and nitrogen control regulatory (NTCA) proteins) that are probable paralogs of CRP that do not bind a cNMP, and several hypothetical sequences from the deduced proteomes of Mycobacterium tuberculosis, Synechocystis PCC6803, C. elegans, and others.
Classification was started by first randomly dividing the cNMP-binding protein superfamily into seven classes, allowing for the six known types of cNMP-binding proteins described in the previous paragraph plus an extra class. PROBE was then used to multiply align the sequences in each class at a purge cutoff of 200. Classifier was applied to these classes and their models for a total of 16 sampling iterations, during which PROBE was called seven times (after every two sampling iterations of Classifier). With each call to PROBE, the purge cutoff value was incremented by 50, up to a maximum of 500. At convergence, seven classes remained with seven distinct models; the final models for the classes were made using PROBE at a purge cutoff of 500.
Classification of this superfamily identified the similarities and differences between classes, identifying motifs unique to individual classes. Figure 2 is a schematic representation of the motifs present in each class. A cNMP-binding domain or β-barrel domain was common to all classes, whereas domains unique to classes included a second cNMP-binding domain, a channel pore motif, and a helix–turn–helix domain. Subtle differences in the cNMP-binding domain were also detected between classes. This allowed the CRP-like proteins (class 5) to form a separate class from the FNR-like proteins (class 4), and also allowed two separate classes to form that each have only a cNMP-binding domain in the class model (classes 6 and 7).
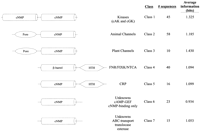
Schematic representations of the cNMP-binding protein classes described in the text. (cNMP) cNMP-binding domain (β-barrel with cNMP contact residues conserved); (β-barrel) β-barrel domain (cNMP contact residues not conserved); (kinase) kinase active site; (pore) pore domain of ion channels; (HTH) helix–turn–helix domain.
Class 1 consisted of the cNMP-dependent kinases from many eukaryotic species, with the class model including regions spanning two cNMP-binding domains. Class 2 contained known cNMP-gated and cNMP-modulated channels, as well as several putative channels of unknown function and regulation from a variety of eukaryotes. The channel class model includes the cNMP-binding domain and a region believed to form the channel pore (Finn et al. 1996). During the last classification iteration, 10 plant channel protein sequences formed a separate class, class 3; the model for this small class also includes a cNMP-binding domain and a pore domain.
Class 4 consisted of sequences from gram-positive as well as gram-negative eubacteria and contained the FNR, FIXK, and NTCA-type prokaryotic transcription regulators. The model for this class spans the regions important for β-barrel formation and a helix–turn–helix. The residues that form hydrogen bonds with cAMP in the β-barrel of CRP were not conserved in this model, consistent with the fact that these proteins do not bind cAMP. The CRP-type prokaryotic regulators formed class 5, containing 16 sequences, most of which are orthologs of E. coli CRP. The model for this class is similar to that of class 4 (encompassing the β-barrel of the cNMP-binding site and a helix–turn–helix), except that those residues that contact cAMP were conserved.
The last two classes contained many hypothetical protein sequences that have entered the database as the result of genome-sequencing projects. The models for these classes included only their cNMP-binding domains and encompassed several subtle variations in sequence conservation in this region between the classes. Class 6 consisted of 23 sequences from prokaryotes, mainly cyanobacteria, as well as lower eukaryotes (C. elegans and fungi). Included in this class was a C. elegans protein that is a putative cAMP–GEF (Kawasaki et al. 1998). The human and rat cAMP–GEFs that were identified recently byKawasaki et al. (1998) were not entered into the nr database at the time of this analysis, and therefore, were not included in the cNMP-binding protein superfamily. Class 7 contained 15 sequences, including many from the predicted proteome of M. tuberculosis, our original query sequence (S. griseus P3), a human esterase, and a C. elegans hypothetical protein.
Nucleotide Cyclases
The seed sequence for the cyclase superfamily was M. tuberculosis Rv1625c (gi‖2113909), a putative adenylyl cyclase (Cole et al. 1998). After partial and duplicate sequences were removed, 163 sequences remained in the superfamily sequence set. The superfamily model consisted of four motifs, shown as sequence logos in Figure3, and encompassed all four residues implicated in catalysis (Liu et al. 1997; Tang and Hurley 1998). An asparagine and an arginine (Fig. 3: motif 4, positions 9 and 13) are believed to be involved in stabilizing the transition state, and two aspartate residues (Fig. 3: motif 1, position 7; motif 2, position 26) likely bind essential metal ions (Mg2+ or Mn2+). Two residues shown to confer nucleotide specificity to eukaryotic cyclases were also included in this model (Fig. 3: motif 2, position 22; motif 4, position 2). Adenylyl cyclases have highly conserved lysine and aspartate residues in these positions, respectively, whereas guanylyl cyclases have highly conserved glutamate and cysteine residues (Tang and Hurley 1998; Tucker et al. 1998).
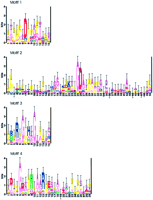
Sequence logos of the nucleotide cyclase superfamily. Sequences were aligned using PROBE with a purge value of 150. The average information at conserved positions is 1.126 bits. The aspartate residues that likely interact with metal ions are located at motif 1, position 7 and motif 2, position 26. The asparagine and arginine residues that are believed to stabilize the transition state are located at motif 4, positions 9 and 13. Two positions that contribute to substrate specificity are located at motif 2, position 22 and motif 4, position 2. Also noted in this superfamily model were several highly conserved glycines, which commonly contribute needed flexibility to the catalytic domains of polypeptides.
This superfamily included nucleotide cyclases from class III as described by Bârzu and Danchin (1994): higher eukaryotic adenylyl cyclases, receptor and soluble guanylyl cyclases, lower eukaryotic adenylyl cyclases, and prokaryotic cyclases. Although many of the superfamily proteins were previously identified cyclases, a significant proportion of the superfamily sequences (>25%) were hypothetical proteins from the deduced proteomes of C. elegans (34 sequences) and M. tuberculosis (15 sequences). Most of theseC. elegans sequences are predicted cyclases (http://www.sanger.ac.uk/Projects/C_elegans/), whereas only five of theM. tuberculosis sequences found are predicted cyclases (http://www.sanger.ac.uk/Projects/M_tuberculosis/).
The classification was started by first randomly dividing the cyclase superfamily into six classes, allowing for the five known types of class III cyclases described in the previous paragraph plus an extra class. PROBE was then used to multiply align each class at a purge cutoff of 200. Classifier was applied to these classes and their models as above—a total of 16 sampling interations, calling PROBE after every two iterations (a total of seven times), and increasing the purge cutoff value by 50 (up to a maximum of 500) with each call to PROBE. At convergence, five classes remained with five distinct models; the final class models were made using PROBE at a purge cutoff of 500. In this example, proteins appeared to form classes due to subtle differences in the catalytic domain, although unique domains were also identified for two of the classes. Figure 4 shows a logo from each of the five classes, illustrating the subtle differences in sequence conservation between classes in the region of the catalytic asparagine and arginine and one of the residues that confers substrate specificity.
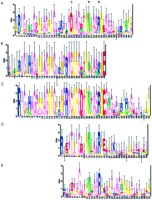
Sequence logos of the catalytic region from each of the nucleotide cyclase classes. The sequences in each class were aligned using PROBE with a purge value of 500 to obtain the class models. The average information at conserved positions for each class model (all motifs) is class 1 (A): 1.234 bits, class 2 (B): 1.366 bits, class 3 (C): 1.458 bits, class 4 (D): 1.264 bits, and class 5 (E): 1.096 bits. The class sequences were scanned with the class models at a purge value of 1000 to make the class logos. The class models are aligned vertically with respect to the catalytic asparagine and arginine. A residue that contributes to substrate specificity is indicated with +, and the catalytic asparagine and arginine are indicated with * (see text). The significance of several highly conserved residues amino-terminal to the catalytic asparagine and arginine in A, B, and C is unknown and is likely because these classes consist mainly of closely related sequences.
Class 1 consisted primarily of the eukaryotic integral membrane adenylyl cyclases, but also included two M. tuberculosissequences, Rv1625c and Rv2435c. The model for this class included only regions from the cytoplasmic domains (C1 and C2) that form the catalytic region of the mammalian adenylyl cyclases. The catalytic asparagine and arginine were well conserved in this class, as was the aspartate that interacts with adenine (Fig. 4A, positions 30, 34, and 23, respectively).
Class 2 contained both the α and β subunits of soluble guanylyl cyclases from many eukaryotic species. The class model consisted of motifs spanning not only the catalytic region, but also sequences in the amino-terminal region of these proteins possibly involved in heme-binding (Hobbs 1997). This class exhibited decreased conservation of the catalytic asparagine and arginine residues (Fig. 4B, positions 33 and 37), consistent with observations that these critical residues are present only on the β subunits, and heterodimerization of the α and β subunits is necessary for activity (Tang and Hurley 1998). Figure 4B shows conservation of the cysteine (position 26) that interacts with guanine, and also an arginine (position 24) that may form hydrogen bonds with guanine via a water molecule (Liu et al. 1997).
Class 3 was the largest class and included known and predicted receptor-type guanylyl cyclases from a variety of eukaryotes. Twenty-two proteins encoded by the C. elegans genome belong to this class, in agreement with the results of Yu et al. (1997), demonstrating the ability of C. elegans to encode a large number of guanylyl cyclases and implying the ability to respond to many sensory stimuli by way of receptor-type cyclases. The class model consisted of motifs spanning the catalytic region and also a kinase-like domain common to the eukaryotic receptor guanylyl cyclases (Wedel and Garbers 1997). The catalytic region exhibited high conservation of the catalytic asparagine and arginine, as well as the cysteine and arginine that likely interact with guanine (Fig. 4C, positions 29, 33, 22, and 20, respectively).
The majority of the prokaryotic cyclases belonged to the remaining two classes. Class 4 contained sequences from Treponema,Stigmatella, mycobacterial, and cyanobacterial species. The class model consisted of motifs spanning only the cyclase catalytic domain. Figure 4D shows that the catalytic asparagine and arginine (positions 12 and 16) were well conserved in this class. Interestingly, class 4 exhibited strong conservation of a threonine residue at position 5 in Figure 4D—a conservative substitution of the cysteine present at this position in eukaryotic guanylyl cyclases. There were also conserved charged residues at positions 1 and 3 in Figure 4D, perhaps corresponding to the arginine believed to interact with guanine via a water molecule in eukaryotic guanylyl cyclases. Guanylyl cyclases have not yet been identified in prokaryotes, although cGMP has been detected in some species (Botsford and Harman 1992).
Class 5 contained several M. tuberculosis sequences, cyclases from several other eubacteria, the receptor-type adenylyl cyclases from protozoa, and the fungal adenylyl cyclases. This class model also consisted of motifs spanning only the cyclase catalytic domain, with conservation of the catalytic asparagine and arginine (Fig. 4E, positions 11 and 15), but a somewhat reduced conservation at the position corresponding to the residue presumed to interact with the substrate purine (Fig. 4E, position 4).
The inclusion of M. tuberculosis Rv1625c and Rv2435c in the eukaryotic adenylyl cyclase class (class 1) prompted us to analyze further these protein sequences. BLAST results of Rv1625c against the SwissProtPlus database using the PAM70 matrix revealed that the most significant hits were to eukaryotic adenylyl and guanylyl cyclases (Fig. 5A). The proteins with alignments having the highest reported bit value scores (soluble guanylyl cyclase subunits from Manduca sexta and Rattus norvegicus) had only a single block of homology with Rv1625c. The human adenylyl cyclase type VIII (CYA8), however, had two separate blocks of homology with Rv1625c (Fig. 5A), making it a highly significant hit when the combined bit value score of the two regions of homology is considered. Alignment of these two sequences (Rv1625c and human CYA8) using the Bayes aligner (Zhu et al. 1998) clearly showed that the regions of homology spanned a contiguous stretch of sequence in Rv1625c, but two separate, distant regions of sequence in human CYA8 (Fig. 5B), thereby encompassing all the critical catalytic residues. The second aligned block shown in Figure 5B included the two metal-binding aspartate residues of human CYA8, and the third aligned block included the catalytic asparagine and arginine residues of human CYA8. Similarly for Rv2435c, BLAST results revealed significant similarity to Rv1625c (one aligned block), as well as several eukaryotic adenylyl cyclases that each had two separate regions of homology with Rv2435c (data not shown). We also constructed a phylogenetic tree to determine how the superfamily of cyclases, in particular Rv1625c, Rv2435c, and mammalian cyclases, may be phylogenetically related (Fig. 6). Indeed, both Rv1625c and Rv2435c grouped with the mammalian adenylyl cyclases on a branch separate from other prokaryotic cyclases.



(See facing page.) (A) BLASTP results given theM. tuberculosis Rv1625c protein sequence as query and SwissProtPlus as the database, using the PAM70 matrix. (B) Bayes alignment of the Rv1625c and human CYA8 protein sequences using the PAM matrices. The four residues implicated in catalysis (two aspartates, an aspargine, and an arginine; see text) are in red and the two residues that confer substrate specificity are in blue.
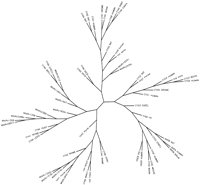
Phylogenetic tree of 60 nucleotide cyclases from our superfamily. All branch lengths were made equal. Cyclases included in the tree are represented by their SwissProt designations; those proteins without a SwissProt name were given a similar designation in lower case, and are as follows: cya9_human is gi‖3138932, cygg_rat is gi‖2833642, cya_xenla is gi‖1514669, cyg_oryla is gi‖1838916, cyg3_manse is gi‖3511175, cya1_mycle is gi‖3097240, cya2_mycle is gi‖3150100, cya_syny3 is gi‖1652963, cya_trepa is gi‖3322767, except for theM. tuberculosis proteins, which are represented by their respective Rv designations.
Phylogenetic Distribution
In the process of identifying and classifying the cNMP-binding protein and nucleotide cyclase superfamilies, we observed that a number of the cNMP-binding proteins did not belong to the known functional classes, and formed new classes with only the cNMP-binding motif in common (classes 6 and 7). We also noted that some species had a large number of nucleotide cyclases. To further investigate the potential functions of these proteins and their phylogenetic distribution, we examined the cNMP-binding proteins and cyclases in our superfamilies, with respect to predicted function, cellular localization, and species (Table 1).
Nucleotide Cyclases and cNMP-Binding Proteins Identified in This study—Cellular Localization and Function
cNMP-Binding Proteins
The proteins from our cNMP-binding protein superfamily were tabulated according to known or predicted function. The majority of the eukaryotic proteins were proteins of known function or shared clear homology to the cAKs and cGKs or the cNMP-regulated channels. The majority of the prokaryotic proteins were also proteins of known function or with clear homology to transcriptional regulatory proteins of the CRP/FNR family. As expected during classification, these proteins formed classes 1–5.
We performed BLAST searches and Pfam domain searches to determine putative functions for the several hypothetical proteins that are members of classes 6 and 7, to reveal whether there may be additional functional classes of cNMP-binding proteins for which there were too few members to form a separate class during our classification procedure. Putative cNMP-regulated functions that were identified were cAMP–GEF, ABC-transporter subunits, antibiotic efflux translocases, and esterases. We also identified protein sequences in eubacteria andArabidopsis thaliana of <200 amino acids that each contain a single cNMP-binding domain spanning virtually the entire protein sequence.
Nucleotide Cyclases
The proteins from our nucleotide cyclase superfamily were tabulated according to known or predicted cellular localization and nucleotide specificity, because there exists a considerable amount of data concerning these characteristics for the eukaryotic cyclases. When unknown, the nucleotide specificity of eukaryotic cyclases was predicted based on the data of Tucker et al. (1998), showing that the lysine/aspartate and glutamate/cysteine residue pairs discussed above are sufficient to confer adenine and guanine specificity, respectively. However, because most of the prokaryotic cyclases were hypothetical sequences arising from genome projects and much less is known about nucleotide specificity among these proteins, the prokaryotic cyclases were all listed in the adenylyl cyclase rows in Table 1, although this should not be considered an assertion about nucleotide specificity. The class I cyclases (Bârzu and Danchin 1994) are unrelated to the cyclases in the class III superfamily, but were included in Table 1 in a separate row.
M. tuberculosis and Synechocystis
Among the prokaryotes, M. tuberculosis H37Rv andSynechocystis PCC6803 each seemed to encode a relatively large number of cNMP-binding proteins and nucleotide cyclases. To compare these species with other prokaryotes, using an unbiased sample set, we compared the prokaryotes (eubacteria and archaea) with completed genome sequences. There are currently 22 completely sequenced prokaryotic genomes, and the predicted proteomes of each of these is available from the National Institute for Biotechnology Information (NCBI). We constructed a sequence set of these 22 proteomes (41,908 total sequences), and scanned (Neuwald et al. 1995, 1997) this set for cNMP-binding proteins and cyclases using our superfamily models. Using this smaller data set, no additional cyclases were detected and just two additional cNMP-binding proteins were detected (sequences that were not in the nr database at the time of the original PROBE search). In addition, we scanned this data set for class I cyclases using a model developed with PROBE by aligning the nine known class I cyclases (sequences from PROSITE motifs PS01092 and PS01093, and the Pfam family PF01295). Table 2 compares the results of these scans with the currently available genome annotation.
Nucleotide Cyclases and cNMP-Binding Proteins in Prokaryotic Species with Available Genome Sequences
DISCUSSION
cNMP-Binding Proteins Superfamily
The majority of sequences in the cNMP-binding protein superfamily classified with one of the known functional classes (kinases, channels, and transcriptional regulators). These functional classes had motifs common to the superfamily (cNMP binding pocket or β-barrel) as well as motifs unique to that class, allowing classification based on those similarities and differences. There were, however, some unexpected findings during this classification.
First, class 1 contained both cAKs and cGKs. cAKs are heterotetramers composed of regulatory subunits (represented in class 1) and catalytic subunits, whereas the cGKs are homodimers that have the kinase active site and cGMP regulatory sites on the same polypeptide. Therefore, we expected that the cAK regulatory subunits would form a separate class from the cGKs, which would contain a kinase domain. This did not happen, however, likely due to the relatively few cGK sequences, which represent only a few species, currently in the database. Such a small group of sequences does not provide enough data to form a separate class using our methods, particularly when they are as highly homologous as the available cGK sequences.
We also expected that the cNMP-gated channels and the cNMP-modulated channels could form sepa
rate classes. Instead, the two channel classes that formed separated a small group of closely related plant sequences (class 3) from other eukaryotic sequences (class 2). It is likely that the sequence signals that distinguish the gated channels from the modulated channels are very subtle, and require more data for identification.
Classes 4 and 5 separated the FNR-type from the CRP-type transcription regulators. These proteins apparently have a similar β-barrel structure (Kolb et al. 1993; Fischer 1994). Both show high conservation of the five glycines that are associated with stabilizing the β-barrel structure. The FNR-type proteins, however, lack the cNMP-contact residues (glutamate and arginine), and are not regulated by cNMPs. Many of these sequences were included in the superfamily purged set (Fig. 1; purge value of 150), resulting in reduced conservation of the glutamate and arginine residues in the superfamily. The FNR-type sequences remained in the superfamily during the jackknife test, indicating that the critical feature of this superfamily was a structural feature (the β-barrel), which extends beyond the cNMP-binding proteins. Interestingly, the FNR proteins fromBacillus subtilis and Bacillus licheniformis were not members of class 4, but members of class 5, indicating the presence of the critical cNMP-contact residues in these proteins. MostBacillus species apparently do not make cNMPs (Kolb et al. 1993); indeed, the genome of B. subtilis does not encode a nucleotide cyclase of the types examined here (see Table 2). Therefore, there has been no selective pressure in these species to drive mutation of the cNMP-contact residues in these FNR proteins, and perhaps too little evolutionary time since acquiring a CRP-like gene for random mutations to have altered these residues.
Of particular interest were the putative cNMP-binding proteins detected in several species that did not classify with any of the known functional classes, and for which no function has yet been predicted. These proteins formed classes 6 and 7, and the proteins within these classes shared only the cNMP-binding domain. One protein was a likely cAMP-GEF from C. elegans. Because this one cAMP–GEF was the only entry in the database at the time, there were not enough data for a class of cAMP–GEFs to have formed during our classification. Using database searches, we determined that there are likely to be additional functional classes of cNMP-binding proteins that have not yet been described, and had too few entries in the database to form a class using our methods. Among these were sequences that appear to contain only the cNMP-binding motif, which spans the majority of the protein sequence (Table 1). It is possible that these proteins are prokaryotic and plant regulatory subunits of cNMP-dependent kinases, regulatory subunits of some other protein complex, or that they function to sequester cNMPs. Also, among “other” functions of prokaryotes in Table 1 were proteins with close homologs in several species (members of the conserved hypotheticals), indicating that there are conserved functions in prokaryotes, yet to be elucidated, that are likely regulated by cNMPs.
Nucleotide Cyclase Superfamily
Whereas previous classifications of nucleotide cyclases have focused on protein topology, cellular localization, and substrate specificity, the classification presented here relied on subtle differences in the residues surrounding the cyclase active site, as well as the presence of unique motifs for the two classes of eukaryotic guanylyl cyclases. The differences in residue conservation of classes 1–3, illustrated in Figure 4A–C, reflected what is currently known about substrate specificity and the catalytic mechanism of the mammalian cyclases, specifically that (1) an aspartate or cysteine residue (marked with + in Fig. 4) contributes to specificity for adenine or guanine, respectively, and (2) that the soluble guanylyl cyclases act as heterodimers, requiring the presence of the catalytic asparagine and arginine (marked with * in Fig. 4) on only the β-subunits (Tang and Hurley 1998).
Our classification identified two classes of prokaryotic nucleotide cyclases: a class that we hypothesize may represent prokaryotic guanylyl cyclases (class 4), and a class that likely represents prokaryotic and unicellular eukaryotic adenylyl cyclases (class 5). The class models for classes 4 and 5 spanned only the cyclase active site; therefore, the simplest explanation for the separation of prokaryotic cyclases into two classes is that the adenylyl and guanylyl cyclases formed separate classes due to differing conservation of the residues conferring substrate specificity. This view may be oversimplistic, however, as it is based on a relatively small number of prokaryotic cyclase sequences available during this study. When compared to the Pfam database, many of the sequences belonging to class 4 also contained various signal tranduction-type domains, including GAF (cGMP phosphodiesterase, adenylyl cyclase, andFhlA), PAS ( p er, a rnt, and s im), FHA (forkhead-associated), and response regulator receiver domains (http://pfam.wustl.edu/; data not shown), suggesting novel modes for regulating the activity of these prokaryotic cyclases.
When tabulating this superfamily, the predicted cellular localization and nucleotide specificity of the eukaryotic cyclases in Table 1conformed to experimental observations. All of the cyclases from multicellular eukaryotes belonged to one of the previously identified groups: (1) integral membrane adenylyl cyclases with 12 transmembrane helices and 2 cytoplasmic domains, (2) receptor-type guanylyl cyclases,and (3) cytoplasmic guanylyl cyclases. The sequences from single-celled eukaryotes were previously identified or predicted adenylyl cyclases (see Table 1) that have been described (Tang and Hurley 1998): protozoan cyclases are receptors, fungal cyclases are peripheral membrane proteins, and Dictyostelium discoideum has one integral membrane and one receptor-type cyclase. Even for the two eukaryotes for which complete genome sequences are available, no cyclases of a previously unrecognized type were detected in the PROBE superfamily.
However, a significant number of proteins among the prokaryotic cyclases were predicted to be integral membrane cyclases, with topology similar to the mammalian adenylyl cyclases (six transmembrane helices and a single cytoplasmic domain), and receptor-type cyclases. Prokaryotic receptor-type nucleotide cyclases have been identified previously only in cyanobacteria (Katayama and Ohmori 1997), and putative prokaryotic integral membrane cyclases have been reported inStigmatella aurantiaca (Coudart-Cavalli et al. 1997) andM. tuberculosis (Tang and Hurley 1998), although how the activity of these enzymes is regulated is unknown.
Archaea
The phylogenetic distribution of both the cNMP-binding protein and cyclase superfamilies indicates an early origin for these proteins, perhaps before the evolutionary separation of the eubacteria from the eukaryotes. Also, the absence of archaeal proteins in our superfamilies suggests either that these proteins were lost from the archaea or evolved after the separation of the archaea from the eubacteria and eukaryotes.
The lack of nucleotide cyclases (class I and class III) and cNMP-binding proteins in the archaea suggests that the archaea either do not use cNMPs as second messengers or produce and bind cNMPs by mechanisms different than those described here. Mechanisms for the production of cNMPs that are unrelated to the class III cyclases have been described. The class I cyclases of the gamma Proteobacteria (Bârzu and Danchin 1994), the class II cyclases of Bacillus anthracis, Bordetella pertussis, and Pseudomonas aeruginosa (Bârzu and Danchin 1994; Yahr et al. 1998), and the novel cyclases of Aeromonas hydrophila (Sismeiro et al. 1998) and Prevotella ruminicola (Cotta et al. 1998) have no apparent sequence similarity to each other or to the class III cyclases, suggesting that at least five different mechanisms have evolved for cNMP production. Sismeiro et al. (1998) reported that some archaeal species encode proteins that are members of a class of cyclases unrelated to the class III cyclases, although nucleotide cyclase activity has not yet been demonstrated in the archaea. Alignment of the nine putative members of this new cyclase class with PROBE (alignment available athttp://www.wadsworth.org/resnres/bioinfo/) revealed several conserved motifs over the length of the sequences, and therefore did not suggest functionally significant regions.
Given the results of Sismeiro et al. (1998) indicating the presence of archaeal cyclases, the absence of archaeal proteins in our cNMP-binding protein superfamily remains puzzling. The available gene annotation indicates the presence of a cNMP-binding motif in theArchaeoglobus fulgidus protein AF0971 (Table 2). To resolve this discrepancy, we performed additional scans of the archaeal proteomes with the PROBE models for the seven cNMP-binding protein classes. AF0971 was detected by the class 4 model (Fig. 2), suggesting that it may be distantly related to the FNR-type regulatory proteins, and is unlikely to bind a cNMP. No other archaeal proteins were detected by these scans. BLAST database searches of the available sequence data also failed to detect any archaeal proteins with homology to other known cNMP-binding proteins, including the extracellular cAMP receptors and novel cAMP-binding proteins of D. discoideum(Grant and Tsang 1990; Firtel 1996), DnaA of E. coli (Hughes et al. 1988), and cGMP-binding cyclic nucleotide phosphodiesterases (Charbonneau et al. 1990). The possibility remains that the archaea have evolved a completely different mechanism than those described here to produce and bind cNMPs.
M. tuberculosis
M. tuberculosis and Synechocystis both appear to have an unparalleled number of putative cNMP-binding proteins, althoughSynechocystis encodes relatively few cyclases. The functions of only half of these cNMP-binding proteins could be predicted by homology (Table 1). The large number of cNMP-binding proteins in bothM. tuberculosis and Synechocystis suggests a previously unappreciated importance of cNMPs to these species and perhaps to other eubacteria.
The M. tuberculosis proteins in the cyclase superfamily were of particular interest for several reasons: the large number (15) ofM. tuberculosis proteins in this superfamily, the presence of predicted cytoplasmic (9), receptor-type membrane bound (1), and integral membrane (5) cyclases, and two M. tuberculosisproteins (Rv1625c and Rv2435c) classified with the multicellular eukaryotic adenylyl cyclases during our classification of this superfamily.
The large number of putative cyclases in M. tuberculosisimplies that this organism may have the ability to sense and respond to many intracellular and extracellular signals through the cNMP second messenger system, perhaps in a manner similar to eukaryotic cyclases.M. tuberculosis encodes a number of putative cytoplasmic cyclases, which could respond to intracellular signals in a manner similar to the eukaryotic soluble guanylyl cyclases (nitric oxide) or the class I cyclases (nutrient availability). M. tuberculosisalso encodes a putative receptor-type cyclase, similar in topology to the eukaryotic receptor guanylyl cyclases, implying the ability to sense an extracellular signal. The extracellular domain of this protein (Rv2435c) has homology to a chemotaxis receptor in Desulfovibrio vulgaris for which the ligand is unknown (Deckers and Voordouw 1996). Also present in M. tuberculosis were putative integral membrane cyclases similar in topology to the multicellular eukaryotic adenylyl cyclases. This raises an interesting possibility that M. tuberculosis could respond to extracellular signals in a manner similar to the mammalian cyclases, by using GTPases (G proteins) or other intermediary proteins in a signal cascade at the inner membrane surface. Considering the large number of M. tuberculosisproteins identified in the cyclase superfamily, it is also a possibility that this organism could use cNMPs as intercellular messengers in a manner similar to D. discoideum. We have not, however, identified any M. tuberculosis proteins with homology to the D. discoideum cAMP receptor proteins.
Although it is not known whether a M. tuberculosis cyclase activity is necessary for pathogenesis, it has been reported that macrophages with ingested mycobacteria have increased levels of cAMP and that phagosome–lysosome fusion is impaired (Lowrie et al. 1975,1979). A link between cAMP levels and pathogenesis has been demonstrated previously—the adenylyl cyclase toxin (ACT) of B. pertussis is able to cause unregulated cAMP production in the host cell, an ability that is necessary for pathogenicity (Gross et al. 1992). In addition, Masure (1993) demonstrated that ACT cyclase activity contributes to intracellular survival of B. pertussisin macrophages, perhaps by inhibiting critical phagocyte activities (chemotaxis and oxidative response) with elevated cAMP levels in the macrophage. Our results, combined with these data, suggest an important role for cNMPs to M. tuberculosis.
Phylogenetic Analysis of the Cyclases
Phylogenetic analysis of the nucleotide cyclases gave similar results as our classification, showing five major groups of cyclases and, in particular, placing the M. tuberculosis proteins Rv1625c and Rv2435c on a branch with the eukaryotic adenylyl cyclases. Our alignment of Rv1625c with a human cyclase further illustrates the surprising degree of sequence similarity between these proteins from such distant organisms. These results suggest a possible horizontal transfer event, an event that could have given an ancient mycobacterium a survival advantage as mycobacterial species were becoming pathogens of eukaryotes. Preliminary sequence results indicate that a Rv1625c ortholog is present in other pathogenic mycobacterial species, supporting this notion that a common ancestor of pathogenic mycobacteria acquired the gene. Unfortunately, there are no current genome sequencing projects for nonpathogenic mycobacterial species, therefore we could not confirm the absence of a Rv1625c ortholog in any of these species to support our hypothesis.
The sequence databases have been expanding rapidly in recent years, and with the currently ongoing genome sequencing projects—>50 prokaryotic genomes, >10 unicellular eukaryotic genomes, as well as multicellular eukaryotic genomes such asH. sapiens, Mus musculus, Drosophila melanogaster, and A. thaliana—these databases will continue to expand ever more rapidly. These genome sequencing projects have produced large amounts of sequence data that are not derived from the traditional hypothesis-driven scientific process. Although these sequence data are provided without evidence of protein function, mutant phenotypes, etc., which come from hypothesis-driven research, they do allow scientists an opportunity to view an organism as a whole, to consider the entire coding capacity of a cell, and conduct research that is data driven. We chose to analyze the cNMP-binding proteins and nucleotide cyclases in the light of the completion of several prokaryotic genomes and two eukaryotic genomes. The genome projects have contributed considerable data to these protein families, and our application of Bayesian protein family classification methods to analyze these growing protein families revealed previously unrecognized, potentially important roles for cNMPs in M. tuberculosis and Synechocystis.
METHODS
Database mining and multiple sequence alignments were performed with PROBE (Liu et al. 1999; Neuwald et al. 1997). PROBE uses a purge procedure based on the BLOSUM62 matrix to remove closely related sequences before creating the alignment model. The PROBE alignment model is “superlocal,” aligning only functionally constrained regions of the proteins and ignoring all other regions. Superfamily members were identified and aligned using PROBE to mine the NCBI nonredundant database (272,992 sequences) using a single seed sequence (the query) to start and a default purge cutoff of 150. Partial and duplicate sequences were removed from the superfamily sequence set and the remaining sequences realigned using PROBE. Alignment of a set of sequences was done by giving PROBE the set of sequences as both the query and the database, and the resulting alignment called the superfamily model. Sequence logos (Schneider and Stephens 1990) of the alignment models were made using WebLogo (http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi).
Classification of the superfamily sequences was performed with the Bayesian sequence Classifier (Qu et al. 1998), a recently developed procedure that iterates between an alignment step, which uses PROBE, and a classification step, which uses the predictive update version of the Gibbs sampler (Liu et al. 1995). Briefly, sequences were divided into an empirically determined number of classes, typically allowing at least 25 sequences per class, and the sequences in each class were multiply aligned using PROBE. During the classification step, each sequence was removed from its class (and class model) one at a time and reassigned to a class in proportion to the posterior probability of class membership. Iteration between alignment and classification continued until convergence. The final class models were then made using PROBE to align the sequences. The initial number of classes need not be equal to the final number of classes—at convergence classes may be empty. This procedure separates a superfamily of proteins into classes, identifying the sequence similarities and differences between classes.
A jackknife procedure was used to detect false-positive members of the superfamilies. After classification of a superfamily, the members of a class were removed from the superfamily, the remaining members of the superfamily aligned using PROBE (with a purge value of 150), and the resulting model used to scan the nr database. A class was considered to contain false positives and discarded if at least one of the class members was not detected by this reduced model (E-value ≤0.01, all BLOSUM scores >200). This procedure was carried out for each class in a superfamily, but no classes were discarded.
The cellular localization of hypothetical proteins was predicted using TMHMM, which uses hidden Markov models to predict transmembrane helices (Sonnhammer et al. 1998;http://www.cbs.dtu.dk/services/TMHMM-1.0/), and TMPRED, which predicts transmembrane helices using weight matrices based on known transmembrane proteins (http://www.ch.embnet.org/software/TMPRED_form.html).
Pairwise sequence alignment was performed with the Bayes aligner (Zhu et al. 1998), a procedure designed to align only those regions that are conserved (local alignment) without having to set a gap penalty or specify a scoring matrix. The two sequences (the query and the data) were aligned using the PAM matrices (PAM 40–300).
Phylogenetic trees were constructed using PHYLIP (Felsenstein 1993). The sequences to be included were multiply aligned with PROBE as described above. The phylogenetic tree was inferred using the protein distances (PROTDIST) and neighbor-joining (NEIGHBOR) methods; the unrooted tree was drawn using DRAWTREE.
Acknowledgments
We thank J. Kwak and K.E. Kendrick for bringing our attention toS. griseus P3 and cNMP-binding proteins, and for helpful discussions. We also thank the Computational Molecular Biology and Statistics Core at the Wadsworth Center for assistance throughout this project and Michael Palumbo for assistance with the jackknife test. This research was supported by National Institutes of Health grant 5RO1-HG0125703 to C.E.L. Data sets and sequence alignments of the superfamilies and classes are available athttp://www.wadsworth.org/resnres/bioinfo/.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL lawrence{at}wadsworth.org; FAX (518) 473-2900.
-
- Received June 2, 1999.
- Accepted December 7, 1999.
- Cold Spring Harbor Laboratory Press








