
Identification, Characterization, and Mapping of Expressed Sequence Tags from an Embryonic Zebrafish Heart cDNA Library
- Christopher Ton1,
- David M. Hwang1,
- Adam A. Dempsey1,
- Hong-Chang Tang1,
- Jennifer Yoon1,
- Mindy Lim1,
- John D. Mably2,
- Mark C. Fishman2, and
- Choong-Chin Liew1,3,4
- 1Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5G 1L5, Canada; 2Cardiovascular Research Center, Massachusetts General Hospital, Charlestown, Massachusetts 02129, USA and Department of Medicine, Harvard Medical School, Boston, Massachusetts 02115, USA; 3The Cardiovascular Genomic Unit, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts 02115, USA
Abstract
The generation of expressed sequence tags (ESTs) has proven to be a rapid and economical approach by which to identify and characterize expressed genes. We generated 5102 ESTs from a 3-d-old embryonic zebrafish heart cDNA library. Of these, 57.6% matched to known genes, 14.2% matched only to other ESTs, and 27.8% showed no match to any ESTs or known genes. Clustering of all ESTs identified 359 unique clusters comprising 1771 ESTs, whereas the remaining 3331 ESTs did not cluster. This estimates the number of unique genes identified in the data set to be approximately 3690. A total of 1242 unique known genes were used to analyze the gene expression patterns in the zebrafish embryonic heart. These were categorized into seven categories on the basis of gene function. The largest class of genes represented those involved in gene/protein expression (25.9% of known transcripts). This class was followed by genes involved in metabolism (18.7%), cell structure/motility (16.4%), cell signaling and communication (9.6%), cell/organism defense (7.1%), and cell division (4.4%). Unclassified genes constituted the remaining 17.91%. Radiation hybrid mapping was performed for 102 ESTs and comparison of map positions between zebrafish and human identified new synteny groups. Continued comparative analysis will be useful in defining the boundaries of conserved chromosome segments between zebrafish and humans, which will facilitate the transfer of genetic information between the two organisms and improve our understanding of vertebrate evolution.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos.BE693120–BE693210 and BE704450.]
The Human Genome Project (HGP) has amassed a vast quantity of sequencing data; over 90% of the human genes have been deposited into GenBank (June 2000). However, functional interpretation of this sequence data has proven more challenging. Much of this work has involved the study of model organisms because functional inferences based on interspecies comparison of sequences have identified implied function of many orthologous human sequences (Makalowski and Boguski 1998).
Recently the zebrafish, Danio rerio, has been recognized as a useful model for the study of development biology and genetics (for review, see Driever and Fishman 1996). One significant advantage of using the zebrafish as a model organism for developmental study is the external development and transparency of the zebrafish embryo. This permits the study of subtle developmental phenotypes in vivo. The zebrafish is also well suited for studies in cardiovascular development because a beating heart is formed and functions within 1 d of fertilization. In addition, the zebrafish embryo does not require blood flow for survival during the first 2 d of development. Thus, zebrafish mutants lacking a circulatory system can still develop normally in the first 2 d (Warren and Fishman 1998) and this allows for studies of mutations that affect the development of the zebrafish heart. Despite all these advantages, the zebrafish suffers from the major drawback of being a new model organism. For example, the number of genes that have been characterized from this species is small compared with other model organisms such as mouse, Drosophila, and Caenorhabditis elegans.
Expressed sequence tags (ESTs) have proven to be a powerful and rapid approach to identify new genes that are preferentially expressed in certain tissue or cell types (Hwang et al. 1997; Liew et al. 1994;Adams et al. 1995). ESTs have also been used for physical mapping, as has been demonstrated in the development of the human and mouse gene maps (Hayes et al. 1996; McCarthy et al. 1997; Deloukas et al. 1998). Currently, the number of zebrafish ESTs in the public databases is still small compared with mammalian sequences, and there are relatively few tissue-specific cDNA libraries.
Mutational screens in the zebrafish have identified several thousand mutations that affect normal development of the embryo (Development, Dec. 1996), including many with essential functions during embryonic heart development (Chen et al. 1996;Stainier et al. 1996). However, the usefulness of these mutations remains limited until the genes responsible for the observed phenotypes are cloned. This is limited in part by a paucity of ordered genes on the zebrafish gene map. Linkage maps based on rapid amplified polymorphic DNAs and microsatellite markers have been produced (Postlethwait et al. 1994, 1999; Johnson et al. 1996; Knapik et al. 1996, 1998; Shimoda et al. 1999). Because linkage mapping requires polymorphic markers for map construction, radiation hybrid (RH) mapping provides a complementary approach to rapidly assign genes and ESTs on the zebrafish map because RH mapping is able to map virtually any marker. Two recent RH maps (Geisler et al. 1999; Hukriede et al. 1999) of more than 3000 markers, genes, and ESTs have dramatically increased the density of the zebrafish gene map and should facilitate the cloning of many identified mutants.
Given the potential and importance of the zebrafish as a model organism for the studies of cardiac development, there is a need for development of EST resources from zebrafish heart cDNA libraries. Here we report the characterization of 5102 ESTs from a 3-d-old zebrafish embryonic heart cDNA library. We also report new map positions for 98 zebrafish ESTs identified in this cDNA library by RH mapping (Table 1) and identification of new synteny groups between zebrafish and human. This EST database represents a new genomic tool for studying aspects of cardiovascular development and disease in the zebrafish and a resource of genes for novel candidate gene discovery.
List of Mapped Zebrafish ESTs
A total of 5102 EST sequences were processed with the TIGR Assembler to estimate the number of unique transcripts represented in the EST set. A total of 359 clusters composed of 1771 ESTs were generated, whereas the remaining 3331 ESTs did not cluster. The number of unique transcripts identified from the zebrafish embryonic heart EST set was therefore estimated at up to 3690.
RESULTS
Overview of ESTs from the Zebrafish Embryonic Heart cDNA Library
A unidirectional cDNA library was constructed from 3-d-old zebrafish embryonic hearts. A total of 5102 random clones were partially sequenced from this cDNA library to generate ESTs. In total, 2937 (57.6%) showed significant identity to known sequences in the nonredundant nucleotide and peptide databases; of these, 946 were zebrafish entries. Another 722 (14.1%) ESTs matched to other ESTs in dbEST but not to any known sequences. The remaining 1418 (27.8%) showed no match to any known sequences and were designated as novel genes (Table2).
Summary of ESTs from the Zebrafish Embryonic Heart
A total of 5102 EST sequences were processed with the TIGR Assembler to estimate the number of unique transcripts represented in the EST set. A total of 359 clusters composed of 1771 ESTs were generated, whereas the remaining 3331 ESTs did not cluster. The number of unique transcripts identified from the zebrafish embryonic heart EST set was therefore estimated at up to 3690.
Known Gene Expression Profile in Zebrafish Embryonic Heart
ESTs matching to known genes were categorized into seven categories on the basis of general functions of the genes (cell division, cell signaling/communication, cell structure/motility, cell organism/defense, gene/protein expression, metabolism, and unclassified) (Adams et al. 1995; Hwang et al. 1997). In total, 1242 unique known genes were represented and the percentage of transcripts in each category was calculated. The largest class of genes represented those involved in gene/protein expression (25.9%). This class was followed by genes involved in metabolism (18.7%), cell structure/motility (16.4%), cell signaling and communication (9.6%), cell/organism defense (7.1%), and cell division (4.4%). Genes lacking enough information to be classified constituted the remaining 17.9% (Table3).
Functional Distribution of Known Genes
Consistent with the high proportions of ESTs involved in gene/protein expression, ribosomal proteins were some of the most abundantly expressed (Table 4). Among other abundantly expressed genes, nine copies of the bone morphogenetic protein 4 (BMP4) were identified. Within the category cell structure/motility, the largest groups of ESTs represented contractile proteins, cytoskeletal proteins, and components of extracellular matrix. The high frequency of these transcripts was not unexpected for the heart, on the basis of our previous experience. However, an unusually high number of keratin proteins (75 clones) and cytokeratin proteins (77 clones) were identified, perhaps due to inclusion of some noncardiac tissues during the isolation of the embryonic hearts.
Most Abundant Genes Expressed in the Embryonic Zebrafish Heart
Comparative Analysis of Gene Expression Profile between Human Fetal Heart and Zebrafish Embryonic Heart
To determine similarities and differences between the two-chambered zebrafish and the four-chambered human heart, we compared proportions of genes in each functional category by using human fetal data fromHwang et al. (1997). Significant differences were detected in five different functional categories. It was found that in the zebrafish embryonic heart, there were significantly fewer transcripts encoding proteins that function in cell division (P < .005), cell signaling/communication (P < .001), and gene/protein expression (P < .001), whereas those involved in cell structure/motility and cell/organism defense were significantly increased (P < .001) relative to human fetal heart (Fig.1; Table 5). Detailed analysis of subcategories found that the decrease in cell division-related transcripts in zebrafish was due to a lower proportion of transcripts representing the general factors of cell division, whereas the decrease in cell/signaling communication was a result of the relative scarcity of identifiable growth factors and hormones in the zebrafish (Table 6). However, the number of transcripts representing effectors/modulators was significantly higher in the zebrafish. This increase could be attributed to a large number transcripts for parvalbumin, a calcium sequesterer detected in fish cardiac muscle (Laforet et al. 1991). Analysis of the cell structure/motility category revealed that extracellular matrix was the only subcategory that showed a significant decrease. However, the number of transcripts representing cytoskeletal proteins was much higher in the zebrafish. This increase was due to the large number of keratin and cytokeratin transcripts present. In the gene/protein expression category, the transcription factors, postranslational modification, ribosomal proteins, and translation factors subcategories all decreased significantly in the zebrafish.
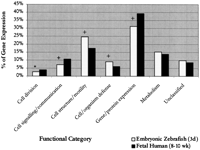
Comparison of relative levels of gene expression between embryonic zebrafish and human fetal hearts. Represented are levels of gene expression in the embryonic zebrafish and fetal human hearts in seven different functional categories. The χ2 test was used to determine statistical significance (*P = .005;+ P = .001).
Relative Levels of Gene Expression in the Embryonic Zebrafish and Fetal Human Hearts
Zebrafish-Human Syntenies
Significantly more ESTs were detected in the cell/organism defense category in the zebrafish, due largely to increases in three subcategories: general homeostasis, carrier proteins, and stress response. Although significant change was not detected in overall levels of transcripts devoted to metabolism, some subcategories exhibited significant changes. Specifically, the nucleotide and transport subcategories showed significant increases, but the sugar/glycolysis subcategory showed decreases. There were also significantly more ADP/ATP carrier proteins and ion-transporting ATPases identified in the zebrafish than in the human heart.
RH Mapping of Embryonic Heart ESTs
Primers were designed for 127 selected ESTs. Of these, 101 (79%) successfully amplified a zebrafish PCR product. Eleven of the primer pairs (9%) failed to amplify a detectable PCR product from zebrafish DNA, and primers for another 8 (6%) ESTs produced Hamster PCR products that could not be clearly distinguished from Zebrafish PCR products. Two primer pairs (2%) were designed for ESTs that are not covered in the hybrid panel (retentions frequency 0%) and primers for 5 (3%) other ESTs produced wrong size PCR products and were discarded. In total, mapping reactions were reproducibly scored for 102 genes represented in the EST set. Of these, 98 (96%) were successfully assigned to single linkage groups (LG), with 23 of 25 groups represented (Table 1). Linkage group 16 contained the most genes (n = 10), followed by LG 7 (n = 8), LG1 (n = 7), and LG6 (n = 6). No genes demonstrated significant linkage to LG21 or LG25 in this analysis (Table 1).
Synteny Analysis
To further analyze the conservation of synteny between zebrafish and humans, we compared positions of the mapped zebrafish ESTs and their human counterparts. Following the method described by Gates et al. (1999), we have identified one new conserved syntenic group between zebrafish and human and added more genes to the previously identified groups. Comparing map positions of zebrafish ESTs and human orthologs identified a new syntenic group belongs to linkage group 16 in zebrafish and chromosome 19 in human and added one to two extra genes to each of five previously identified groups (Table 6).
DISCUSSION
The generation of ESTs has proven to be a useful and rapid means to identify and isolate large numbers of expressed sequences (Adams et al. 1992, 1993; Hwang et al. 1994, 1995; Liew et al. 1994). Although extensive EST-based resources exist for human and other mammalian models such as mouse and rat, the EST database for the zebrafish presently contains approximately 100,000 ESTs and is still being developed (Gong et al. 1997; Gong 1999). In this report, we characterized the transcriptional profile of 3-d-old embryonic zebrafish hearts by generation of 5102 ESTs. Clustering of 5102 ESTs estimated the maximum number of unique genes represented in this set at 3690. Because this analysis was performed on 5′ end sequences that may arise from multiple nonoverlapping segments of the same gene, the true number of unique genes is almost certainly lower.
Of known gene matches, a number of genes thought to be involved in cardiogenesis were identified in the data set. These included nine copies of BMP4, which has been found to be involved in the regulation of left-right asymmetries of the zebrafish heart (Chen et al. 1997;Schilling et al. 1999). Other important factors known to regulate cardiogenesis were also identified, including homeobox transcription factors Nkx2.3/2.5, Mef2A/2C, and atrial natriuretic factor.
Although comparative analyses of DNA sequences have been performed between model organisms and humans (Koop 1995; Makalowski et al. 1996;Makalowski and Boguski 1998), little attention has been paid to studying the patterns of gene expression variations between model organisms and humans on a global scale. Understanding similarities and differences between identical tissues in different species is essential in establishing “synexpression” data sets, defining groups of genes that share a similar functional pathway (Niehrs and Pollet 1999). To investigate similarities and differences in gene expression profiles in the developing heart between zebrafish and humans, we analyzed relative levels of expression of genes with related functions. Despite limitations of comparing these two data sets at different stages of development, these findings provide us with a first look at global differences in overall physiological status between the two-chambered zebrafish and the four-chambered human heart, though for the most part, the analysis was too small to reliably reveal differences in the transcription of specific genes. Nevertheless, the results of this analysis suggest several interesting differences in patterns of expression. For example, the high frequency of transcripts detected in the cell/organism defense category in the zebrafish may indicate differences in homeostatic requirements between zebrafish and human hearts. A proportionally high number of heat shock cognate 70 transcripts (hsc70) was detected in the zebrafish heart, with 31 ESTs representing this gene (0.6% of all ESTs). This represents a significant increase in proportion of hsc70 expression over human fetal heart (0.1% of all ESTs; Hwang et al. 1997). Heat shock cognate 70 functions as a chaperone and is known to protect cells against apoptosis (Hohfeld 1998). Heat shock proteins can also be induced by environmental stress. Unlike human fetuses that develop in a stable environment in utero, fish embryos develop externally and it is plausible that the increased levels of hsc70 in the zebrafish embryonic heart may serve a protective role during embryonic development in the face of a potentially changing environment.
Beyond analysis of expression profiles, one immediate application of this EST resource is as a substrate for RH mapping. Recent reports have dramatically increased the number of mapped zebrafish markers, genes, and ESTs (Geisler et al. 1999; Hukriede et al. 1999). Here, we present mapping results for an additional 102 ESTs identified from our library that should further facilitate the identification of zebrafish mutant genes with essential functions during zebrafish embryonic development (Chen et al. 1996; Stainier et al. 1996).
Comparative analysis of map positions between zebrafish and human has identified that gene orthologs that are syntenic in mammals are also syntenic in zebrafish (Postlethwait et al. 1998). This discovery of extensive sharing of chromosome segments between zebrafish and humans has practical significance to the HGP. For example, synteny between zebrafish and humans will enable researchers to identify human ortholog from a gene's position in the zebrafish genome. Reciprocally, and more importantly, the phenotype of a zebrafish mutation can suggest function for the human gene (Postlethwait and Talbot 1997). However, before any conclusive characterization can be made about this conservation, more detailed analyses of these conservations are needed to further define the boundaries of conserved chromosome segments and the extent to which gene order is maintained between zebrafish and human. This information would be particularly useful in identifying candidate genes for positional cloning analyses. It is anticipated that the continuing development of a dense zebrafish map will markedly increase its utility and facilitate the transfer of genetics information between the zebrafish and human.
This collection of 5102 ESTs provides us with a preliminary view into the gene expression profile of the zebrafish embryonic heart. The identification of many genes known to be involved in cardiogenesis suggests that the generation of ESTs is an excellent method for identifying additional genes with essential roles in heart development. Further integration with mapping data of these zebrafish ESTs will provide a richer resource for identifying candidate genes for the several thousand mutants that affect zebrafish development. Construction and characterization of cDNA libraries from additional stages of development, with comparison of gene expression profiles between libraries, should provide further valuable insights into the molecular mechanisms of heart development and disease.
METHODS
RNA Isolation
Total RNA was isolated from 3-d-old zebrafish embryonic heart samples by the method described by Chomczynski and Sacchi (1987). Tissues were homogenized and extracted twice with acidic guanidinium isothiocyanate-phenol-chloroform. The poly(A)+ RNA fraction was isolated by oligo-dT cellulose chromatography (Pharmacia). Purity and RNA integrity were assessed by absorbance at 260/280 nm and agarose gel electrophoresis.
cDNA Library Construction
Libraries were constructed in the λZAP Express vector (Stratagene) according to the manufacturer's protocols. First-strand cDNA was synthesized with an XhoI-oligo(dT) adapter-primer. After second-strand synthesis and ligation of EcoRI adapters, cDNA was digested with XhoI, generating cDNA flanked byEcoRI sites at 5′ ends and XhoI sites at the 3′ ends. Digested cDNAs were size-fractionated with Sephacryl S-500 spin columns and ligated into the λZAP Express vector predigested with EcoRI and XhoI. The resulting concatomers were packaged by using Gigapack Gold packaging extracts. After titration, aliquots of primary packaging mix were stored in 7% DMSO at –80°C as primary library stocks, and the remainder was amplified to establish stable library stocks.
Partial Sequencing of 5′ Ends of cDNA Inserts
Plaques were picked randomly and eluted into SM buffer. Phage eluates (5 μL) were directly used for PCR reactions (50-μL final volume). Reaction mixtures contain 5 μL of 10X Taq buffer, 125 μL of each dNTP, 10 pmol each of forward primer (5′-GCCAAGCTCGAAATTAACCCTCACTAAAGGG-3′) and reverse primer (5′-CCAGTGAATTGTAATACGACTCACTAT AGGGCG-3′) and 1 U of Taq polymerase. The thermal cycle profile consisted of an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 45 sec, 57°C for 30 sec, and 72°C for 3 min, and a final extension step of 72°C for 3 min. After agarose gel electrophoresis to determine the purity and concentration, 2 μL of PCR products were used directly for cycle sequencing by using the AmpliCycle Sequencing Kit (Perkin-Elmer) and 5 pmol of Cy5 labeled modified T3 primer (5′-GAAATTAACCCTCACTAAAGG-3′). The conditions for cycle sequencing were as follows: 94°C for 2 min, followed by 35 cycles of linear amplification (94°C, 30 sec; 50°C, 15 sec; 72°C, 1 min for 20 cycles and 94°C, 30 sec; 72°C, 1 min for 15 cycles). The reactions were stopped by addition of 0.5 v/v loading buffer (95% formamide, 20 mmol/L EDTA, 10 mg/mL blue dextran). Sequencing reactions were loaded onto 6% acrylamide gels and electrophoresed with A.L.F. and A.L.F. Express DNA sequencers (Pharmacia) (Hwang et al. 1995,1997).
Bioinformatics
Sequence search analysis of all ESTs against the nonredundant GenBank/EMBL/DDBJ nucleotide, nonredundant GenBank CDS translation/PDB/SwissProt/PIR/PRF peptide, and dbEST databases were performed with the BLAST algorithm (Altschul et al. 1990; Gish and States 1993) on a Unix platform (Sun Microsystems). Assignment of putative identities for ESTs required a minimum P value of 10–10. ESTs with known gene matches were categorized into different functional groups according to categories described in Hwang et al. (1997). Relative levels of gene expression were computed by summing the number of ESTs matching to that particular gene and dividing the sum by the total of ESTs that match to known genes (Hwang et al. 1997). The combined 5102 ESTs were clustered on the basis of sequence similarity by using TIGR Assembler (Fleischmann et al. 1995). Parameters were set so that ESTs were connected together only with a minimum of 95% nucleotide identity in an overlap region of 40 nucleotides. GenBank accession nos. of the Zebrafish Embryonic heart ESTs are AI353073-AI354214; AI616386-AI618739; AI618836-AI618858;AW453485-AW455194. Further clone information can be found on the Internet at URL www.tcgu.med.utoronto.ca.
Preparation of DNA Templates for 3′ End Sequencing
The cDNA clones were excised in vivo from the λZAP Express vector by using ExAssist/XLOLR helper phage system (Stratagene) before sequencing. Phagemid particles were excised by coinfectingEscherichia coli XL1-BLUE MRF′ cells with ExAssist helper phage. Excised pBluescript phagemids were used to infect E. coli XLOLR cells and selected by using kanamycin resistance. Single colonies were grown overnight in LB-kanamycin and DNA purified by using Qiagen plasmid purification kits. Purified DNA was then used for sequencing of 3′ ends.
Radiation Hybrid (RH) Mapping of cDNA Clones
A 94-hybrid zebrafish RH panel was purchased from Research Genetics. 3′-end sequences of each EST were used to design PCR primers with the assistance of the Williamstone Enterprises Primer Design program (http://www.williamstone.com). Primers were generally 20-bp long and were chosen to generate PCR products of 100–300 bp and a Tm range of 58–60°C. Primer pairs that showed high complementarity to each other or similarity to repeat sequences were discarded. ESTs for which no satisfactory primer pair was found were not used. Names, symbols, and primer sequences are summarized in Table1. Each primer pair was pretested for specificity with zebrafish and hamster genomic DNA (Research Genetics). Primer pairs that gave a specific zebrafish product were used to screen the RH panel.
PCR amplification was performed in 10-μL reaction mixtures containing reaction buffer, 2mM each dNTP, 0.05 U Taq polymerase, 4 pM each primer, and 5 ng each hybrid. The thermal cycle profile consisted of an initial denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 1 min, 58°C for 1 min, and 72°C for 1 min, and a final extension step of 72°C for 10 min. PCR products were separated by gel electrophoresis in 2% agarose with 0.5X TBE, and photographed on a UV transilluminator.
Each primer pair was tested in duplicate and positive products were scored. In case of discrepancies (positive on one plate but negative on the other), the band(s) were rescored. Retention profiles were submitted to the Max Planck Institute (Tubingen, Germany) for analysis by SAMapper 1.0 (Geisler et al. 1999).
Statistical Analysis
Analysis of differences in expression levels between zebrafish and human genes was performed by using 2606 and 10,854 unique genes respectively, with ESTs from the mitochondrial genome excluded from calculations. The expected number of zebrafish ESTs present in each functional category/subcategory was calculated based on the frequency of the observed number of ESTs in the fetal human heart cDNA library. By using the same method for identifying differentially expressed genes from EST-based expression profiles as described in Hwang et al. (2000), the statistical significance of the deviation of observed EST profiles from expected was tested with the χ2 test. For each category, the χ2 value was calculated by summing the χ2 value for that category with the χ2value calculated from the sum of the remaining category/subcategories. Statistical significance of the deviation from expectations was tested by the χ2 value with one d.f. The thresholds of significance were established at *P = .005 and+ P = .001. The statistical significance of deviation between the two sample sizes was confirmed by using another method for assessing significance of gene expression profiles as described in Audic and Claverie (1997) (http://igs-server.cnrs-mrs.fr).
Phylogenetic Analysis
Following the method described by Gates et al. (1999), each EST sequence was searched against the protein database at NCBI by using the BLASTX program (Altschul et al. 1990). Mammalian sequences that showed significant similarity to the zebrafish EST were retrieved. These sequences were then multiply aligned and neighbor-joining trees were constructed by using CLUSTALX (Thompson et al. 1997). A zebrafish EST is orthologous to a human gene if it appears as a sister group on the dendrogram. The locations of human gene loci were taken from Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/); the Genome Database (http://www.gdb.org/gdb), and The Human Gene Map (http://www.ncbi.nlm.nih.gov/genemap99/).
Acknowledgments
We are grateful to Jack Liew for oligonucleotide synthesis, Wei Wei for assistance with automated sequencing, Robert Geisler and Gerd-Jörg Rauch for calculating map positions on the RH map and to everyone at the Cardiac Gene Unit for technical assistance. This work was supported by the Medical Research Council of Canada. C.T. is a recipient of a Heart and Stroke Foundation of Canada Traineeship. D.M.H. is a recipient of a Hunt Estate M.D./Ph.D. Studentship. A.A.D. is a recipient of a Heart and Stroke Foundation of Canada Traineeship. J. Y. is a recipient of a Heart and Stroke Foundation of Ontario Summer Student Scholarship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL cliew{at}rics.bwh.harvard.edu; FAX (617) 975-0995.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.154000.
-
- Received June 30, 2000.
- Accepted October 10, 2000.
- Cold Spring Harbor Laboratory Press








