
HAPPY Days for the Dictyostelium Genome Project
HAPPY mapping allows construction of a map of a piece of DNA without the need to clone it, thus avoiding many potential errors and artifacts (Dear and Cook 1993; Piper et al. 1999). The method is based on random DNA breakage and determination of linkage. The power and potential of the technique are well illustrated in the article by Konfortov et al. (2000, this issue). The authors use HAPPY mapping to construct a physical map of chromosome 6 ofDictyostelium. They map 300 sequence-tagged sites to the 4-mb chromosome, giving an average marker spacing of 14 kb. Some of the mapped sites fit with previously mapped sites, determined from an ordered YAC library (Kuspa and Loomis 1996), but many do not. Therein lies the rub, because these discrepancies posed a substantial, hidden threat to the Dictyostelium genome project. This map and the map now being constructed of the whole Dictyostelium genome (P. Dear, pers. comm.) have put the project firmly back on course. We will explain the original “unhappy” situation and what lessons can be learned for other genome projects. But first, we will give some information on the Dictyostelium genome project.
Why Sequence the Dictyostelium Genome?
To answer this question, it is necessary to answer the larger question: What is Dictyostelium? For a long time, botanists and zoologists both laid claim to it, but analysis of conserved protein sequences now indicates an evolutionary relationship much closer to animals than to plants and one that places it in the crown group of organisms (Loomis and Smith 1990; Baldauf and Doolittle 1997). This conclusion fits very well with the biology of theDictyostelium cell and the regulatory pathways that control its developmental cycle (Parent and Devreotes 1996; Baldauf and Doolittle 1997; Aubry and Firtel 1999). It is highly motile and undertakes chemotaxis, pinocytosis, and phagocytosis in a manner very similar to human leukocytes. These properties have made it a very attractive model system in which to study the many processes that animal cells perform but yeast cells do not. It is a good model for these processes because the molecular machinery inDictyostelium closely resembles that of animal cells, but its genetics are much better. Also it provides unique opportunities to study evolutionarily conserved signaling pathways controlling cell-fate decisions in a multicellular organism. These include pathways that function via STAT transcription factors (Williams 1999), β-catenin, and GSK3 homologs—signaling molecules of great importance in animal development. The Dictyostelium genome project, as it rapidly moves along, is making Dictyostelium an even more attractive experimental system because it is revealing many more conserved proteins just crying out for gene disruption, overexpression, suppressor analysis, and so forth (Kay and Williams 1999).
The Genome Project: A Progress Report
The project is international and division of labor is on a chromosome basis. The chromosomes are purified by PFGE. There are six chromosomes, and all but chromosomes 4 and 5 are resolvable. The DNA is used to prepare shotgun libraries in a plasmid vector, and the inserts are sequenced from both ends. The chromosomes that can be resolved are estimated to be ∼60% pure, and the contaminants that are present in a preparation of any one chromosome derive from all the other chromosomes though not, of course, equally. Therefore, simultaneously with building up coverage on their target chromosome(s), each group is also contributing to a whole-genome shotgun. This affords very good coverage, several-fold the genome size but with a bias in favor of chromosomes 2 and 6, the first two chromosomes to be assigned for sequencing. The total genomic sequence data can be searched on a number of sites (seehttp://genome.imb-jena.de/Dictyostelium/project.html,http://www-biology.ucsd.edu/others/dsmith/dictydb.html,http://dictygenome.bcm.tmc.edu/). Intermediate assembly of the shotgun reads has been performed and has yielded almost 5000 contigs of >2 Kb that cover 17.5 Mb of the 34-Mb genome (http://www.sanger.ac.uk/Projects/D_discoideum/). The genomic sequence data are supplemented by an EST project (http://www.csm.biol.tsukuba.ac.jp/cDNAproject.html), in which a consortium of Japanese groups have sequenced almost 5000 individual (unique) cDNAs from the slug stage of development (Fig.1). In an organism estimated to express only 8000–10,000 genes, this is a highly significant number, which will simplify the gene prediction/annotation.
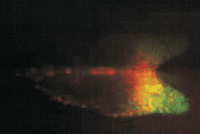
This is a living Dictyostelium slug containing ∼20,000 prestalk cells in the front one-fifth and 80,000 prespore cells in the rear four-fifths. There are also scattered prestalk-like cells in the prespore region. There are two kinds of prestalk cells in the prestalk region that are defined by their use of different promoter elements from the ecmA gene. In the slug shown in this image (D. Dorman, T. Abe, J. Williams, and C. Weijer, unpubl.) the two promoter regions were coupled to different GFPs and the fluorescence is from pstA cells (red) in the tip, pstO cells (green) behind them, and a region where the two cell populations overlap (yellow).
The HAPPY Map Dimension
The strategy for the Dictyostelium genome project is based on that used for the Plasmodium falciparum, the malarial parasite. It was adopted because Dictyostelium andPlasmodium have very similar genomic organizations. The coding regions are islands of relative GC-richness (∼30%–40%) in a sea of very low GC-richness (∼10%–20%). This is both good news and bad news. The good news is that the coding regions can be readily distinguished from the introns and intergenic regions; that is, gene prediction is relatively easy. The bad news is that the assembly process is difficult; the long runs of A and T cause cloning, sequencing, and computer alignment problems. These problems are compounded because ∼10% of the Dictyostelium genome is composed of dispersed repetitive elements of one kind or another. Thus, a whole-genome shotgun approach, in which the assembly process is left to the computer, seems unlikely to work; there would be almost as many gaps in the final sequence as there would be genes.
The strategy being used for Plasmodium is to separate chromosomes by PFGE and perform a whole-chromosome shotgun. A parallel shotgun “skim” (i.e., a shotgun with a low number of reads) is performed on a minimal tiling set of YAC clones (i.e., a set of minimally overlapping YAC clones) that cover the chromosome. The YAC skims produce a set of anchor points that have a known sequence and an approximately known location. In the case of the Dictyosteliumproject, in which only 60% of shotgun reads actually derive from the desired chromosome, such an approach is highly effective, as the YAC skim reads “pull in” the reads that derive from the target chromosome. Moreover, the other reads are not wasted because the collaborating groups interchange primary sequence data and clones and the data are used to help achieve greater coverage of all the chromosomes.
There is, however, one essential precondition to the above approach. The overlapping YAC clones that comprise the map must be correctly assigned; A YAC clone assigned to the wrong chromosomal position will pull in reads from the wrong chromosome, and assembly will be incorrect. This is a dangerous Achilles' heel, as the generally used methods of constructing YAC libraries do not prevent multiple inserts from being cloned: YAC clones can be chimeric, with regions of the insert being derived from different chromosomes or noncontiguous regions of the same chromosome. This will lead to mismapping and most probably explains why the HAPPY map and the YAC map of chromosome 6 display many discrepancies. Now that over one-half of the YACs are known to be incorrectly positioned on the map, a new minimal tiling set can be selected using correctly mapped YACs. This can be done using PCR primer pairs with a known HAPPY map position to test whether particular YACs do indeed derive from the chromosomal positions to which they were originally assigned.
Summary
Without the HAPPY map, the collaborators in the genome project would have found assembly to be extremely difficult, and theDictyostelium genome sequence would perhaps have been left highly incomplete. With the HAPPY Map the YAC clones can be remapped and the original YAC skim strategy followed. In conclusion, this method has already made one community very happy and seems sure to make its mark in many other genome projects.
Footnotes
-
↵3 Corresponding author. E-MAIL ; FAX 44-1382-345823.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.166100.
- Cold Spring Harbor Laboratory Press








