
DNA Profiling of B Chromosomes from the Yellow-necked Mouse Apodemus flavicollis (Rodentia, Mammalia)
Abstract
Using AP-PCR-based DNA profiling we examined some structural features of B chromosomes from yellow-necked mice Apodemus flavicollis. Mice harboring one, two, or three or lacking B chromosomes were examined. Chromosomal structure was scanned for variant bands by using a series of arbitrary primers and from these, informative bands were selected. The selection criteria used were the ability to differentiate between individuals of the species, to detect markers common for both A and B chromosomes, and, importantly, to differentiate between A- and B-chromosome sets. In addition to primers, profiling conditions were found to be critical for meeting the selection criteria. Primers and analysis conditions that demonstrated structural characteristics unique to the B-chromosome set are described. These characteristics included variant bands as qualitative parameters and altered electrophoretic band intensities as quantitative distinctions estimated by integration of densitometric profiles of electrophoretograms. B chromosome-specific molecular markers are easy to detect by AP-PCR-based DNA profiling in the presence of a full set of A chromosomes. Models for the origin of yellow-necked mouse B chromosomes are discussed in the context of presented data.
B chromosomes are additional chromosomes that are not essential for the viability of an organism and are therefore often described as accessory or supernumerary chromosomes. They represent an extremely variable class of chromosomes with regard to their morphology, phenotypic effects, and modes of transmission. B chromosomes (Bs) fail to pair with any member of the A-chromosome set (As) during meiosis, although they may pair and form chiasmata among themselves. They are characterized by non-Mendelian assortment, and their occurrence throughout the plant and animal kingdom is widespread (Jones and Rees 1982).
A variety of models for the evolutionary origin of Bs have been designed without reaching a consensus. The most widely accepted view is that Bs are derived from the As (Jones and Rees 1982). They could be derived from the leftover centromere from A fusions (Patton 1977), from polysomic As, from the amplification of the paracentromeric region of a fragmented A (Keyl and Hagele 1971), or from fragments from trisomic pairing (Amos and Dover 1981). Most Bs seem to have originated from the autosomes of their host species (Jamilena et al. 1994a, 1995; Houben et al. 1996, 1997; Stark et al. 1996; Peppers et al. 1997), but there are examples of sex chromosome-derived Bs (Lopez-Leon et al. 1994; Sharbel et al. 1998). Some recent findings support the idea that Bs could be derived from the As of a closely related species in interspecific crosses (Sapre and Deshpande 1987; Schartl et al. 1995; McAllister and Werren 1997).
A conceptually different hypothesis suggests that Bs might be a prokaryotic counterpart of an independent replicon analogous to a bacterial plasmid (Brockhouse et al. 1989). This suggests the absence of gross sequence homology between the two sets of chromosomes, which is in contrast to the findings in plant and mammalian genomes (McQuade et al. 1994; Jamilena et al. 1994a, 1994b, 1995; Stark et al. 1996). It is generally accepted that Bs share extensive homology with the A set and that some Bs have repetitive sequences distinct from those present in the A complement (Beukeboom 1994). A reasonable mechanism, known as Muller's ratchet (Green 1990), allows sequences that are not under selective pressure to evolve rapidly with each generation envisioned (analogous to the click of a ratchet). According to this mechanism, Bs should initially be largely euchromatic and homologous to As from which they arose and should become more heterochromatic and show less homology over time. This hypothesis gained some support in the finding that newly formed Bs in maize were more heterochromatic with each succeeding generation (Peeters et al. 1985). Accumulation of repeat sequences or insertion of transposable elements may represent mechanisms for B differentiation from their homolog progenitors (J.P.M. Camacho, T.F. Sharbel, and L.W. Beukeboom, in prep.). Alternative explanations for this phenomenon are conceivable as the process of heterochromatization is still not understood. For more details on B origin and evolution, see J.P.M. Camacho, T.F. Sharbel, and L.W. Beukeboom (in prep.).
One of the peculiarities of the genus Apodemus is the frequent occurrence of animals possessing Bs. In yellow-necked miceApodemus flavicollis two kinds of Bs exist. They are either smaller than the smallest chromosomes of the basic complement (Kral et al. 1979; Zima 1984) or of the same size as the five smallest chromosomes of the standard set (Vujos̆evic andZ̆ivković 1987). During meiotic division they appear as univalents or bivalents (Sablina et al. 1985; Vujos̆evic et al. 1989). The fact that the same number of Bs was found in bone marrow and in testicular tissue, together with the fact that no differences in the distribution of Bs between males and females were found, leads to the conclusion that neither the mechanism of their elimination nor accumulation is operating during meiotic division (Vujos̆ević 1992). The application of differential staining shows homology in the distribution of G and C bands between As and Bs (Vujos̆ević and Z̆ivković 1987), which means that differences between these chromosomes are beyond the resolution of cytological analysis. According to their morphology and responses to G and C banding, it can be hypothesized that Bs in A. flavicollis represent direct products of the A set. Most likely they were derived from polysomy of small autosomes that had not yet been followed by true heterochromatization (Vujos̆ević andZ̆ivković 1987). They may prove to be determinants of population dynamics in yellow-necked mice (Blagojević and Vujos̆ević 1995).
Our former findings suggest that Bs of A. flavicollisoriginate from the basic A set via some abnormal genomic event(s), the mechanism of which remains to be elucidated. This fact inspires an alternative option to approach the problem, namely the analogy with genomic abnormalities involved in the process of malignant transformation. The analogy is rather limited and plausible only from the methodological point of view. Microsatellite instability, encountered in some major human malignancies, is characterized by hundreds of thousands of somatic mutations in simple repeated sequences scattered over the genome (Perucho 1996). Such heritable chromosomal abnormalities on the somatic cell level were discovered by the application of DNA fingerprinting by arbitrarily primed PCR (AP-PCR). This unbiased methodology allows for molecular karyotyping of somatically acquired genomic abnormalities in anonymous regions of the genome. Thus, comparing related genomes, whereby one is a derivative of the other emerging via an undefined and abnormal genomic event, makes AP-PCR analysis of As and Bs a reasonable approach.
This report presents the application of the AP-PCR approach of genome analysis and aims to improve the understanding of the nature and the origin of Bs in yellow-necked mice. Our experimental design is analogous to the comparative analysis of normal and malignant tissue except that tissues of yellow-necked mice without Bs and animals containing Bs are compared. The results obtained demonstrate molecular markers specific for Bs. These specificities include both qualitative and quantitative changes in the genome of yellow-necked mice harboring Bs.
RESULTS
Comparative Restriction Enzyme Analysis of Yellow-necked Mouse Bs
Comparative restriction enzyme analysis was the focus of our initial effort to detect molecular features characteristic of Bs. Genomic digests of 0B and +B DNA (1B, 2B, and 3B) were compared. In this study, six restriction enzymes were examined carefully, as described in Methods. Each enzyme yielded a number of different bands that are indicative of repetitive DNA sequences (Flavell 1982). However, no additional bands or differences in band intensity were observed in specimens with Bs. This result indicates that highly repetitive DNA sequences on Bs that are detectable by restriction analysis are indistinguishable from those on As. It did not make any difference if they had 4- or 6-bp recognition sites or if they were methylation sensitive (data not shown). Preliminary characterization of additional restriction enzymes yielded similar results but was discontinued because it seemed likely that no informative data would be obtained.
AP-PCR Analysis of Yellow-necked Mouse Bs
DNA isolated from yellow-necked mice with and without Bs was analyzed by AP-PCR profiling. Reaction conditions were optimized, primers selected, and reproducibility verified as described in Methods. Figure 1 depicts the analysis with E8A primer. The presented analysis included two animals without Bs (0B) and two animals with two Bs (+B). Their profile was tested at two template concentrations, 50 and 250 ng, which, as expected, yielded minimal differences in respective profiles exemplified by minor variations in band intensities. These experiments exclude the possibility that template quality and interexperiment variability affect the interpretation of the DNA profile. The gel image demonstrates that this type of analysis differentiates between individuals and thus displays the cardinal feature of the DNA profile analysis. Additionally, some bands are characteristic for the species, being common to all analyzed animals, including both 0B and +B individuals. Importantly, some electrophoretic bands are present in DNA profiles of all +B individuals and in none of the analyzed 0B animals. Consequently, these bands are regarded as molecular features unique to Bs. This specificity was observed in at least three individual +B animals from the respective groups harboring one, two, or three Bs. The indicated variant electrophoretic bands represent either sequences unique to the Bs or repeated sequences with variable numbers of repeats.
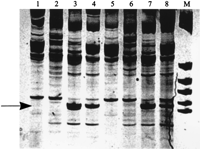
Amplification profile of yellow-necked mice Apodemus flavicolliswith 2Bs (lanes 3,4,7,8) and without Bs (lanes1,2,5,6) using E8a p53 primer. Amplification was performed at two template concentrations: 50 ng (lanes 1–4) and 250 ng (lanes5–8). B-specific bands are indicated by the arrow. (M) Size marker: pBR 322 digested with HaeIII.
The same type of experiment was performed using c-myc1 primer, and the results obtained are shown in Figure 2. In this case, different electrophoretic patterns yielded the same basic information. It was reassuring to observe the same information obtained with a different AP-primer, under a modified set of experimental conditions, and in a different set of experiments. Thus, DNA profiling analysis appears to be a reliable tool for the assignment of molecular specificities to Bs in a sample where As predominate. To enhance the visualization of the observed features, the lower portion of Figure 2illustrates densitometric scans of the selected parts of the gel image. The scans compare DNA profiles of 0B and +B animals.
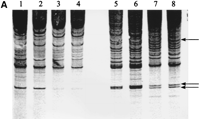
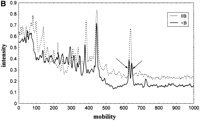
(A) AP-PCR profile of yellow-necked mice A. flavicollis with Bs (lanes 5–8) and without Bs (lanes 1–4) using c-myc1 amplimer. Profiles were tested at two template concentrations: 50 ng (lanes1,3,5,7) and 250 ng (lanes2,4,6,8). B-specific bands are indicated by arrows. (B) Densitometric scan of 0B and +B amplification profile. Arrows indicate B-specific c-myc1 markers.
AP-PCR-based DNA profile analysis of Bs also yielded information about +B DNA specificities that can be classified as quantitative. Figure 3shows the results of amplification with E6S p53 amplimer. The gel image clearly illustrates highly significant differences in band intensities obtained with DNA profiling of +B samples (individuals with 2Bs). This difference in band intensity is not the consequence of multiple, closely spaced bands because a gel underloaded with the same sample yielded a single sharp band with the same electrophoretic mobility (data not shown). Table 1 shows the results of volume integration of peaks differing in intensity, confirming the visual inspection of the gel images (Fig.3). This amounts to a 200%–300% gain in B specific peak intensity. This quantitative difference of correspondent band intensities is yet another molecular feature of Bs.
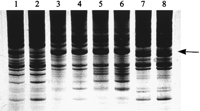
AP-PCR profile of yellow-necked mice A. flavicollis with Bs (lanes 3–6) and without Bs (lanes1,2,7,8) using E6S p53 amplimer. AP-PCR was done at two template concentrations: 50 ng (lanes1,3,5,7) and 250 ng (lanes2,4,6,8). Band intensity changes are indicated by the arrow.
Quantitative Comparison of DNA Profiles with 2B and 0B Chromosomes
DISCUSSION
In our attempt to contribute to an understanding of the nature of mammalian Bs, restriction enzyme analysis did not prove to be very informative. Using a larger assortment of restriction enzymes researchers have attempted to gain a better understanding of Bs in a variety of organisms but have been similarly unsuccessful (Jamilena et al. 1994b; Stark et al. 1996). Nevertheless, a brief discussion is justified because of lack of data for mammalian Bs. The change in the abundance and repeat number of highly repetitive sequences can be detected by comparing 0B and +B genotypes following restriction digestion and gel electrophoresis. Isoschizomer enzymes with distinct sensitivity for the methylation status of their recognition sequences may provide information about the bulk methylation differences among genomes. This type of analysis was successful in the case of some plant genomes (Sandery et al. 1990) where variant B-specific repetitive sequences were reported. In our data, however, 0B and +B yellow-necked mouse genomic DNA digests were indistinguishable, suggesting identity in terms of the abundance and repeat number of highly repetitive DNA. However, it is important to recognize the inherent limitations of the method, namely that restriction analyses are incapable of detecting alterations in dispersed repetitive sequences and are limited to tandem repeats. A possibility remains that a much larger array of restriction enzymes might yield information about repetitive sequences that differentiate between the two sets of chromosomes.
For these reasons we employed a different approach—AP-PCR-based DNA profiling. Technically, it is similar to work reported previously (Stark et al. 1996) for the analysis of Bs from the maize genome. Their RAPD analysis was similar to screening for polymorphisms among bulked sergregant populations or near-isogenic lines. However, our approach was inspired by the fundamental fact that almost all models for the origin of Bs agree that they are derived from the A set, whereby one is a derivative of the other emerging via abnormal genomic event(s). The mode of B chromosome origin and its differentiation is, as a rough outline, analogous to acquired genomic abnormalities that underlie carcinogenesis associated with microsatellite instability. The analogy between cancer and Bs is quite limited in that they having in common only the facts that the genomes are related and that one is derived from the other via aberrant chromosomal maintenance. However, these facts suffice to justify the use of the methodology that proved to be successful in the analysis of microsatellite instability (Perucho 1996). For this reason we compared 0B and +B genomes in the same manner as normal and malignant tissues were examined, in an attempt to detect quantitative (aneuploidy-like) and qualitative (mutation-like) alterations.
The unbiased nature of AP-PCR profiling allows for the screening of anonymous regions of a genome without any prior knowledge of its structure (Welsh and McClelland 1990; Williams et al. 1990) and provides information about two distinct types of DNA alterations. These alterations represent the accumulation of changes in DNA sequence (qualitative changes) that manifest as mobility shifts in the banding pattern, while amplifications or deletions of existing chromosomal material (quantitative changes) are evident as altered band intensities in the banding pattern. Observed changes should be regarded cautiously as semiquantitative and semiqualitative due to the competitive nature of AP-PCR in which sequence context may play an unpredictable role. This situation may be a serious problem for simple to moderate patterns but not for complex patterns. Unfortunately, the former are preferred due to simplicity of interpretation. Because the profile is the result of a competition between many PCR products, the problem may appear with very simple profiles in the analysis of similar but nonidentical genomes. For this reason, we used a profile pattern with >10 prominent PCR products of moderate complexity (McClelland and Welsh 1994). Following the necessary precautions for reproducibility and reliability of DNA profiling analysis, we compared 0B and 1B, 2B, and 3B yellow-necked mouse genomes. Results obtained suggested that AP-PCR DNA profiling can distinguish Bs in the context of the full set of As. We considered only the B-specific bands in DNA profiles that showed invariable molecular features associated with Bs, appearing in all of the +B animals and in none of the 0B animals, regardless of individual or geographical variation. Observed molecular markers for Bs, including 1B, 2B, and 3B genotypes, delineate both qualitative and quantitative changes associated with the presumed evolvement from the A set. DNA profiles differentiate between individual animals and depict species-specific markers in addition to +B markers. The observed quantitative changes, exemplified by the electrophoretic bands with significantly increased intensity, suggest amplification of anonymous regions (but +B specific) of the yellow-necked mouse genome in the process responsible for the origin of Bs. B-specific, increased band intensity is suggestive of gross similarity (or near identity) among 1B, 2B, and 3B genotypes. We observed no quantitative differences in B-specific bands in 1B, 2B, and 3B animals. We propose that in this case, an arbitrary primer amplifies highly favored genomic loci (i.e., repetitive sequences). The relative contributions of the template concentration, exemplified by factors 2 or 3 in 2B and 3B animals, is low compared to 0B and 1B samples with presumed repetitive sequences. Contribution of repetitive sequences to the template concentration may vary by orders of magnitude. Therefore, template concentrations differing by a factor of 2 or 3 may not be detectable (McClelland and Welsh 1994).
Similarly, qualitative changes, apparent as new, B-specific, electrophoretic bands, suggest mutational events involved in evolution and/or maintenance of Bs. Importantly, no qualitative differences were observed among 1B, 2B, and 3B genotypes, implying sequence similarity, at least within the limits of this analytical procedure. Mutational events associated with Bs are hardly surprising, bearing in mind their abnormal biology (i.e., non-Mendelian assortment) and less stringent evolutionary requirement for integrity of Bs (their dispensable nature) in comparison to the basic set and the presumed aberrant nature of the genomic process through which they arise. To go from suggestion to conclusion, regarding the evolutionary significance of described genetic markers specific for Bs, one must remember the competitive nature of AP-PCR and the possible effects of sequence context. For this reason it seems prudent to test an array of related mapping procedures for the analysis of anonymous genome regions and examine if their different underlying principles would lead to the same conclusions. Some of these procedures, tec-MAAP (Caetano-Anolles et al. 1993), double-stringency fingerprinting (Matioli and de Brito 1995), and hairpin primers (Caetano-Anolles and Gresshoff 1996) are currently being tested.
METHODS
Animals
All specimens examined were collected in their natural habitat from three different localities in Serbia using Longworth traps. A. flavicollis with and without Bs were identified by cytogenetic analysis. Chromosome preparations were made directly from bone marrow using standard techniques. Staining of chromosomes was done with slightly modified schedules of Seabright (1971) for G banding andArrighi and Hsu (1971) for C banding. A minimum of 30 spreads from each specimen was analyzed to confirm the exact number of Bs. Excised liver material from animals of known B number was frozen and stored at −70°C before DNA extraction.
DNA Extraction
Nuclear DNA was isolated from the livers of yellow-necked mice harboring Bs and those without Bs, as described by Maniatis et al. (1982). DNA was prepared from 7 animals with one B, 5 with two Bs, and 4 with three Bs (+B) as well as from 10 animals without Bs (0B).
Restriction Enzyme Analysis
Genomic DNA was digested with the following restriction endonucleases: EcoRI, HindIII, HpaII,MspI, MboI, and PvuII, according to Maniatis et al. (1982). The digests were resolved on 0.8% agarose gels in 1× TBE electrophoresis buffer at 1 V/cm overnight at 4°C.
Arbitrarily Primed PCR
The isolated genomic DNA was amplified by AP-PCR. Twenty primers were tested for the ability to generate informative fingerprints. Optimization of AP-PCR reactions was done for each primer according toCobb (1997). Primers that yielded reproducible fingerprints were used for detection of B-specific sequences. Each experiment included the analysis of two template concentrations (50 and 250 ng in the final volume of 25 μl) for each individual to exclude artifacts arising from impurities in DNA preparations. Three primers produced informative polymorphisms differentiating samples carrying Bs and specimens without Bs. Primer sequences were forward primer for exon 8 of the p53gene (E8A) (Sakai et al. 1992), forward primer for exon 6 of thep53 gene (E6S) (Sakai et al. 1992), and forward primer for the c-myc gene (c-myc1) (Abok-Ellela et al. 1996). Optimized reaction conditions were as follows: AP-PCR with primer E8A was performed with 50 and 250 ng of genomic DNA, PCR reaction buffer (Fermentas), 0.4 mm of each of the dNTPs, 2.5 mmMgCl2, 5 μm amplimer, and 1 unit ofTaq DNA polymerase (Fermentas) in a final volume of 25 μl. The reaction profile was: (94°C for 4 min), 4 cycles at low stringency conditions (94°C for 1 min; 45°C for 2 min; 72°C for 2 min), 35 cycles at high-stringency conditions (94°C for 1 min; 64°C for 1 min; 72°C for 2 min), and a final extension (72°C for 5 min). Amplification with E6S primer was performed with 50 and 250 ng of genomic DNA, PCR reaction buffer, 0.2 mm of each of the dNTPs, 2.5 mm MgCl2, 3.5 μm amplimer, and 1 unit of Taq DNA polymerase in a final volume of 25 μl. The reaction profile was identical to that described above except that the annealing for the high-stringency reaction was at 60°C. AP-PCR with c-myc1 primer was performed using 50 and 250 ng of genomic DNA, PCR buffer, 0.2 mm of each of the dNTPs, 2 mm MgCl2, 2 μm primer, and 1 unit of Taq DNA polymerase in a final volume of 25 μl. The reaction profile was denaturation (94°C for 4 min), 4 cycles at low stringency conditions (94°C for 1 min; 45°C for 2 min; 72°C for 2 min), 30 cycles at high stringency conditions (94°C for 1 min; 65°C for 1 min; 72°C for 2 min), and a final extension (72°C for 5 min). Optimization of the reaction also included the search for conditions that yielded profiles of moderate complexity to simplify the analysis (McClelland and Welsh 1994). The AP-PCR products were separated on 8% nondenaturing polyacrylamide gels and visualized by silver staining. Gel images were obtained using Gel Documentation System GDS 8000 SW (TIFF format, Ultraviolet Product Limits, UK), printed on transparent foil, and scanned by densitometer (LKB, Pharmacia XL), which provided superior resolution. Data obtained were evaluated statistically and quantified by STATISTICA (Windows, release 4.5, StatSoft, Inc.) and MD ImageQuant (v. 3.3, Molecular Dynamics).
Reproducibility
Problems with the reproducibility of AP-PCR have been a matter of concern (Meunier and Grimont 1993; McClelland and Welsh 1994). In our case, occasional irreproducibilities were found due to template quality, where an additional round of purification solved the problem. Template carry over was monitored routinely by systematic incorporation of a no-template reaction in each set of experiments. Day to day variation was found only with respect to band intensities. This variability was <15% (±5%) as estimated by integration of densitometric scans.
Acknowledgments
This work was supported by the Ministry of Science and Technology of Serbia, contract no. 03E02.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL nikolata{at}ibbi.ibiss.bg.ac.yu; FAX 381-11-761 433.
-
- Received January 5, 1999.
- Accepted November 3, 1999.
- Cold Spring Harbor Laboratory Press








