Abstract
Haplotype assembly is the problem of reconstructing the combination of alleles on the maternally and paternally inherited chromosome copies. Individual haplotypes are essential to our understanding of how combinations of different variants impact phenotype. In this work, we focus on read-based haplotype assembly of individual diploid genomes, which reconstructs the two haplotypes directly from read alignments at variant loci. We introduce Ralphi, a novel deep reinforcement learning framework for haplotype assembly, which integrates the representational power of deep learning with reinforcement learning to accurately partition read fragments into their respective haplotype sets. To set the reward objective for reinforcement learning, our approach uses the classic reduction of the problem to the maximum fragment cut formulation on fragment graphs, in which nodes correspond to reads and edge weights capture the conflict or agreement of the reads at shared variant sites. We train Ralphi on a diverse data set of fragment graph topologies derived from genomes in the 1000 Genomes Project. We show that Ralphi achieves lower error rates at comparable or longer haplotype block lengths over the state of the art for short and long reads at varying coverage in standard human genome benchmarks.
Haplotype assembly, also known as genome phasing, is the problem of reconstructing the sequence of alleles at sites of genetic variation of each individual chromosome copy in a genome. The specific configuration of variants across homologous genome regions allows us to differentiate between cis variants present on the same chromosome molecule and trans variants present across homologous chromosomes. This information is required to discover compound heterozygous variants, which occur when both homologous copies of a gene contain variants at different positions with a potentially deleterious impact on the function of both gene copies and which play a key role in numerous diseases, such as cancer (De Rosa et al. 2000; Rahner et al. 2008; Miller and Piccolo 2021), Charcot–Marie–Tooth neuropathy (Lupski et al. 2010), ataxia–telangiectasia (Dörk et al. 2004; Piane et al. 2016), and deafness (Welch et al. 2007). As such, the accurate reconstruction of haplotypes is vital to our interpretation of genetic variation and its role in disease (Browning and Browning 2011; Tewhey et al. 2011). Haplotype assemblies are also essential to the study of population structure, genetic diversity, mechanisms of inheritance, and genome evolution (Douglas et al. 2001; Green et al. 2010; Adey et al. 2013; Brinton et al. 2020).
Numerous approaches have been developed to date for haplotype reconstruction. Population-based statistical approaches (Browning and Browning 2007; Loh et al. 2016; Delaneau et al. 2019) infer haplotypes by leveraging observed patterns of genetic variation in a reference panel (e.g., large-scale genotype data sets from The 1000 Genomes Project [The 1000 Genomes Project Consortium 2015] or the UK Biobank [Bycroft et al. 2018]). On the other hand, read-based approaches assemble individual haplotypes using the overlap of read alignments at heterozygous variant loci. Because of sequencing errors, however, the problem of read-based haplotype assembly is computationally intractable. Numerous formulations for this problem have been proposed to date (Panconesi and Sozio 2004; Levy et al. 2007; Bansal and Bafna 2008; Duitama et al. 2010; Aguiar and Istrail 2012; Berger et al. 2014, 2020; Kuleshov 2014; Martin et al. 2016; Edge et al. 2017; Yu et al. 2021; Lin et al. 2022). Among these, some of the most popular definitions include the minimum error correction, minimal fragment removal, and minimal single-nucleotide polymorphism (SNP) removal optimization objectives (Rizzi et al. 2002; Panconesi and Sozio 2004; Duitama et al. 2010). Because these formulations are NP-hard and also hard to approximate (Cilibrasi et al. 2007; Bonizzoni et al. 2016), popular approaches often employ fixed-parameter tractable algorithms (by fixing the read coverage or number of errors as parameters) (Martin et al. 2016; Beretta et al. 2018) and heuristics to solve the original problem (Edge et al. 2017) or reduce the problem space through downsampling (Martin et al. 2016). However, although heuristic-based methods can scale to large problem instances, expert-driven heuristics cannot tractably model the complex sources of variability inherent to this problem domain (e.g., the nonuniformity of read coverage and variant density within the genome and across different populations or read length and error profiles of different sequencing platforms).
Learning-based approaches, on the other hand, can detect complex patterns in high-dimensional data, which are too challenging to discover and define manually. Recently, several deep learning methods have been proposed for the problem of haplotype assembly in polyploid species and viral quasispecies (Ke and Vikalo 2020, 2022; Xue et al. 2022). For example, NeurHap (Xue et al. 2022) formulates haplotype phasing as a graph coloring problem on read-overlap graphs and uses a graph neural network (GNN) to learn color assignments, whereas CAECseq (Ke and Vikalo 2022) uses convolutional auto-encoders to cluster reads into haplotypes. These approaches, however, have been evaluated only on small or partial genomes and read data sets (with most benchmarked data sets containing a few hundred to a few thousand reads and variants); as such, their performance on a full human genome has not been demonstrated.
In this work, we introduce Ralphi, a novel framework for haplotype assembly based on deep reinforcement learning (DRL), which offers the ability to learn combinatorial optimization strategies for high-dimensional inputs. Briefly, in reinforcement learning (RL), an agent is trained to make decisions in a given environment that maximize its reward through trial and error. DRL integrates the representational ability of deep learning with the trial-and-error-based optimization of RL to enable operations on high-dimensional state spaces. It has been shown to achieve state-of-the-art results in a wide range of combinatorial optimization problems, including the traveling salesman, minimum vertex cover, and maximum cut (Khalil et al. 2017). To the best of our knowledge, DRL has never been used for haplotype assembly. In Ralphi, we train a DRL agent to partition read fragments into two haplotype sets while optimizing the maximum fragment cut (MFC) objective proposed by Duitama et al. (2010), which involves solving the NP-hard weighted max-cut problem (Bansal and Bafna 2008; Duitama et al. 2010; Edge et al. 2017) on read-based graphs. Importantly, this approach does not require ground-truth haplotype assignments. Ralphi leverages a graph convolutional neural network (GCN) to embed fragment graphs and an actor-critic RL model to learn the read-to-haplotype assignment algorithm based on the MFC reward. We used a large set of fragment graph topologies derived from genomes from a diverse set of populations included in the 1000 Genomes Project to train Ralphi. To demonstrate the generalizability and adaptability of our learning-based approach to multiple sequencing platforms, we generated Ralphi models for both Oxford Nanopore Technologies (ONT) long reads and Illumina short reads. We demonstrate that Ralphi predominantly improves the accuracy of the reconstructed haplotypes compared with the state of the art, while maintaining high phasing block lengths across several standard benchmark data sets from both short and long read sequencing platforms and different genome coverage regimes.
Results
Overview of Ralphi
At a high level, Ralphi consists of two key modules (Fig. 1): (1) fragment graph construction from read alignments and variants, which generates the input to the learning-based framework, and (2) the DRL framework, which partitions graph fragments into two haplotypes by (1) embedding fragment graphs using a GCN and (2) iteratively assigning each node in the graph to one of the haplotypes using the actor-critic RL algorithm and the MFC reward.
Overview of the Ralphi framework for haplotype assembly. As an example, a fragment graph is constructed from a set of six DNA fragments covering seven SNPs (the allele containing a sequencing error is shown in red), with negative agreement edges shown in blue, and conflict edges, in red (the thickness of the edge corresponds to edge weight). Fragments are partitioned into final haplotypes through an iterative process wherein the agent assigns a node to a haplotype in each step based on the prediction given by the actor-critic network operating over the fragment graph embedding. The final haplotypes are assembled by computing the consensus of the fragments in each partition.
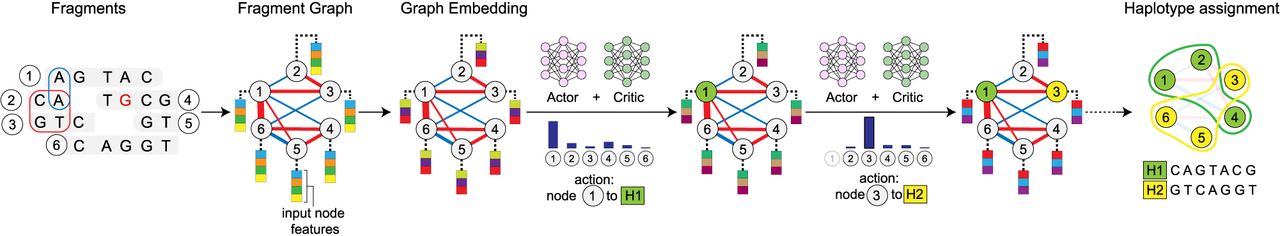
Briefly, in a fragment graph, nodes correspond to DNA fragments, the edges represent the overlap of fragment alignments at variant loci, and the edge weights capture the conflict (positive values) or agreement (negative values) of alleles covered by each fragment. Note, fragments may correspond to a single read alignment or to multiple alignments (e.g., in the case of paired-end Illumina reads). Only fragments that cover two or more variants are informative for phasing and stored in the graph. A cut in a fragment graph represents an assignment of fragments to each haplotype. Intuitively, higher-weight cuts will separate fragments with a higher number of conflicting alleles, which are likely to pertain to different haplotypes.
Given fragment graphs as input, Ralphi learns to assign each node in the graph to one of the two haplotypes such that its assignments maximize the cut value across the two final haplotype sets. For each graph, it incrementally selects one node at a time and assigns it to one of the two haplotypes. Ralphi uses the popular actor-critic RL paradigm (Konda and Tsitsiklis 1999), which involves two networks: (1) the actor, which learns which actions the agent should take (i.e., the policy), and (2) the critic, which learns the value of different actions. Ralphi uses a GCN to capture the graph topology and the state of the node assignments after each action taken by the agent. Each node in the graph is associated with a feature representing its current haplotype assignment (i.e., unassigned, assigned to haplotype 1, or assigned to haplotype 2). Additionally, we explored using other node features to capture various properties of the graph topology (e.g., node degree), fragments (e.g., number of covered variants), and problem context (e.g., how many nodes have already been assigned). We found that Ralphi achieved the best results when betweenness centrality was included as a node feature, which encodes the fraction of shortest paths passing through a given node.
Training data sets
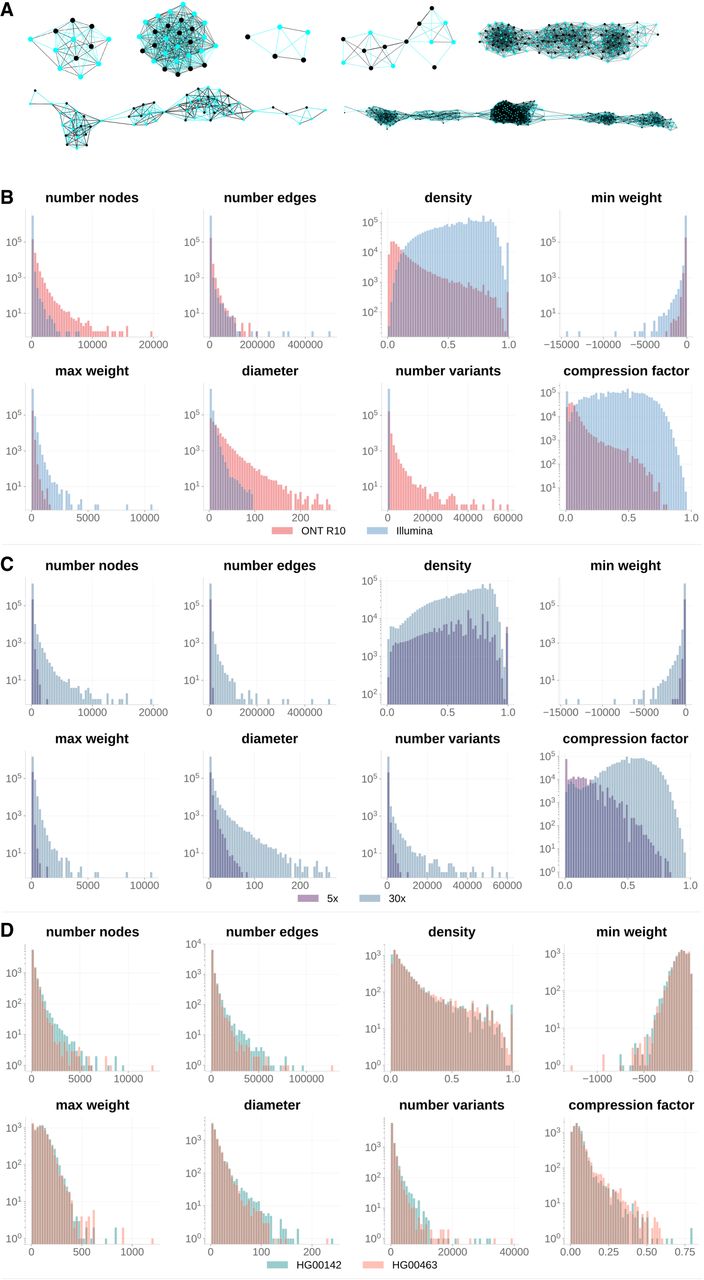
We generated a diverse set of fragment graph topologies for training, aimed at capturing the variability in genome structure (e.g., variant density within and across genomes in different populations), read depth, sequencing error rates, and read lengths. To that end, we selected the following 10 genomes from The 1000 Genomes Project (Byrska-Bishop et al. 2022): HG00234, HG00250, HG00627, HG01598, HG02047, HG03046, HG03166, HG03388, HG04225, and NA11932, which come from diverse populations including the British, Southern Han Chinese, Kinh Vietnamese, Gambian Mandinka, Esan, Mende, and Telegu. We incorporated the variants of each genome into the GRCh38 reference and generated synthetic Illumina short reads and synthetic ONT long reads at varying coverage and error rates from these genomes (see Methods). Additionally, we included real Illumina data sets for each of these 10 genomes and real ONT R10 reads for six additional genomes that have been sequenced on the ONT platform (HG00142, HG00263, HG00277, HG00326, HG00372, and HG00463) at varying coverage. This procedure yielded 3,147,214 graphs for short reads and 184,958 graphs for ONT reads. Figure 2A highlights the diversity of topologies captured in the resulting data set. We computed key topological features (e.g., graph size, density, and diameter) for the resulting graphs shown in Figure 2 B and C (which capture the distributions of fragment graph properties computed for all generated fragment graphs—stratified by technology and coverage); Figure 2D (which displays the distribution of graph properties for two distinct sample genomes derived from real ONT reads); and Supplemental Figure S1 (which includes a larger set of genomes selected for training). We then applied graph sampling strategies to balance across different properties, obtaining a training data set with 267,456 graphs for short reads and 175,048 graphs for long reads. We trained Ralphi first on short-read graphs and then fine-tuned the model on ONT graphs.
Overview of fragment graph characteristics. (A) Examples of fragment graph topologies. Negative and positive edge weights are in black and blue, respectively; node color indicates haplotype membership selected by Ralphi. Distributions of fragment graph properties (note that “compression factor” is defined as 1 − [number of nodes]/[number of fragments]) stratified by technology (B), coverage (C), and genome (D).
Evaluation setup
We evaluated the performance of Ralphi on the NA12878 and HG002 standard benchmark genomes using publicly available Illumina, ONT (R10), and Pacific Biosciences (PacBio) (HiFi) data sets. We compared Ralphi to the popular state-of-the-art phasing methods WhatsHap (Martin et al. 2016), HapCUT2 (Edge et al. 2017), and LongPhase (Lin et al. 2022) developed for long reads (note that we used LongPhase only for ONT and PacBio comparisons). We also included RefHap to directly compare Ralphi's learned algorithm to the heuristic algorithm developed for the same MFC objective (unfortunately only partial metrics are available for RefHap because it did not finish running on all the benchmarks during the course of several months). Supplemental Notes 3 and 4 list the parameters used to execute each tool and their versions. We could not include a comparison with the existing deep-learning methods NeurHap (Xue et al. 2022) and CAECseq (Ke and Vikalo 2022), because the available code does not operate with standard phasing inputs and outputs and requires custom model training. We used the Ralphi model trained only on short reads with Illumina data and the model fine-tuned with ONT reads for both ONT and PacBio data. To assess the effect of coverage on phasing performance, we downsampled the original read data sets to a depth of 30×, 15×, 10×, and 5×. We compare the following key standard phasing metrics to assess performance: switch error rate, mismatch error rate, and the AN50 score, which assess the accuracy and contiguity of the reconstructed haplotypes, respectively (Methods) (Edge et al. 2017).
NA12878 benchmark
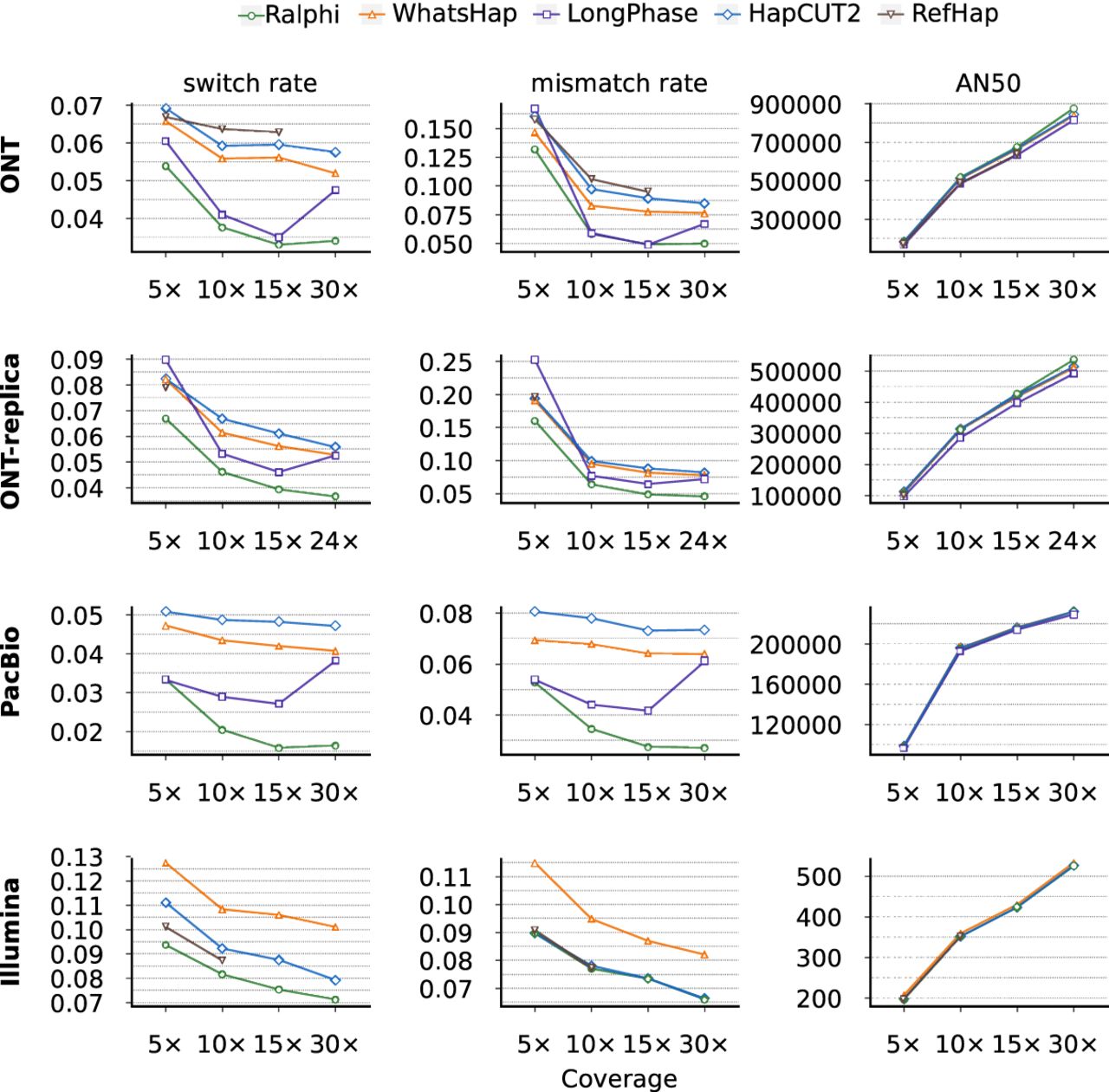
For the NA12878 benchmark, we used phased high-confidence variants from the Illumina Platinum Genomes call set (Eberle et al. 2017) as ground truth. We obtained Illumina 150 bp paired short reads from the 1000 Genomes Project high-coverage NYGC release (Byrska-Bishop et al. 2022), ONT long reads (R10.4.1 chemistry) from the ONT Open Data project (https://labs.epi2me.io/dataindex/), and PacBio HiFi reads from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Vollger et al. 2023). Given the variability in read profiles (read length and quality) of ONT sequencing, we also downloaded a second (replica) ONT data set from the Garvan Institute Long-Read Sequencing Benchmark Data platform (Gamaarachchi et al. 2022) to evaluate performance on the same genome but with different read data sets (please note that the coverage of the BAM file available for this data set was 24×). Figure 3 and Supplemental Figure S2 (with results computed only on Chromosome 1 and Chromosome 20, which were withheld during training) show that Ralphi achieves lower error rates, compared with other methods, across most settings in this benchmark, while maintaining or improving on the AN50 haplotype block contiguity scores; notably, it achieved the highest AN50 at higher coverage with ONT reads. To evaluate the impact of variant selection on performance, we additionally executed Ralphi without any variant filters. Supplemental Figure S4 compares the performance of LongPhase (default mode) and Ralphi (with no variant filters). To ensure that both methods operate on a comparable set of variants, we executed Ralphi in this setting only on the variants phased by LongPhase. It can be seen that Ralphi achieves lower error rates and higher AN50 scores across all coverage regimes as compared to LongPhase.
Performance evaluation on the NA12878 benchmark. Note that switch and mismatch error rates are a percentage.
HG002 benchmark
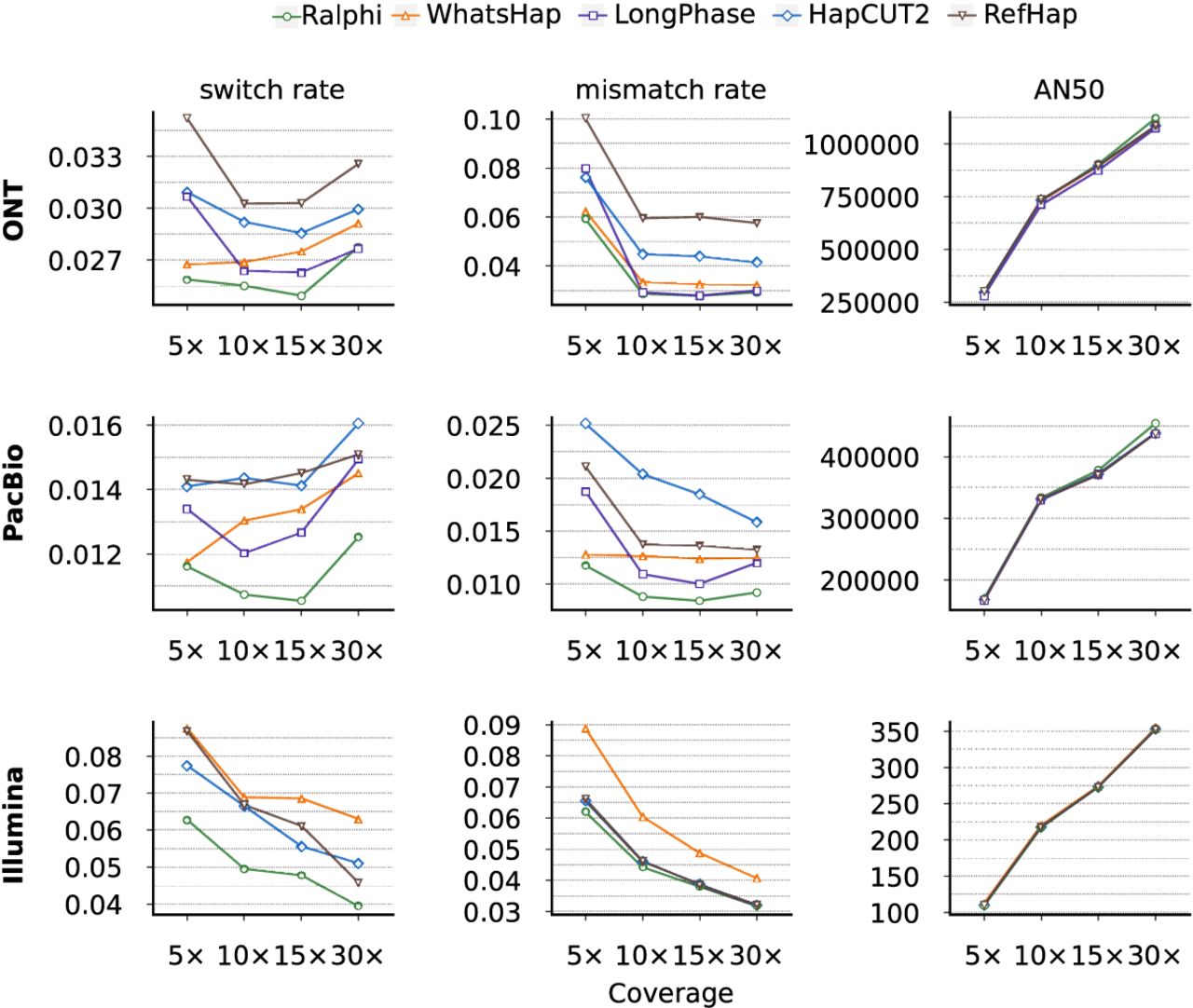
For the HG002 benchmark, we used phased variants from the 4.2.1 GIAB release (Zook et al. 2020) as the ground truth. We obtained 250 bp paired Illumina short reads from GIAB (Jarvis et al. 2022), ONT long reads (R10.4.1 chemistry) from the AnVIL workspace (Kolmogorov et al. 2023), and PacBio HiFi reads from the PacBio cloud (Nurk et al. 2022). Figure 4 and Supplemental Figure S3 (with results computed only on Chromosome 1 and Chromosome 20, which were withheld during training) show that Ralphi similarly achieved lower error rates, compared with other tools, across most settings in this benchmark, and a higher AN50 at higher coverage for ONT and PacBio reads.
Performance evaluation on the HG002 benchmark. Note that switch and mismatch error rates are a percentage.
Runtime evaluation
We evaluated the runtime of Ralphi and other methods on Chromosome 1 of the HG002 genome benchmark (for which all Refhap results finished computing). All our experiments were performed on an AMD Ryzen Threadripper PRO 3995WX system with 516 GB of RAM. We report the runtime of each tool on a single core (all the steps of Ralphi, including the model, were run on a single CPU thread) for short reads and long reads across all coverage regimes in Table 1. LongPhase has the fastest runtime on ONT and PacBio reads, whereas HapCUT2 is the fastest tool on Illumina reads. Ralphi's runtime is generally comparable to WhatsHap, being slower at several coverages on ONT reads and faster at certain coverages on PacBio and Illumina reads. However, although Ralphi is slower than WhatsHap at 30×, it is phasing the full read set, whereas WhatsHap downsamples input data sets to 15× by default. Optionally, Ralphi also provides read downsampling (using WhatsHap's downsampling routine), which can significantly reduce its runtime at 30×. We also show Ralphi's total peak memory usage (the maximum resident set size) at each coverage. Because Ralphi's model is very small, Ralphi is able to achieve a competitive runtime and a low memory footprint even when running with a single thread.
Runtime (min) of each tool on Chr 1 of the HG002 genome for ONT, PacBio, and Illumina reads
| ONT | PacBio | Illumina | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tool | 5× | 10× | 15× | 30× | 5× | 10× | 15× | 30× | 5× | 10× | 15× | 30× |
| HapCUT2 | 0.93 | 1.83 | 2.66 | 4.97 | 0.75 | 1.44 | 1.96 | 4.65 | 0.24 | 0.51 | 0.70 | 1.55 |
| LongPhase | 0.23 | 0.41 | 0.65 | 1.36 | 0.28 | 0.36 | 0.66 | 1.20 | — | — | — | — |
| RefHap | 0.58 | 1.61 | 6.22 | 92.98 | 1.03 | 2.69 | 8.36 | 167.63 | 0.71 | 0.96 | 1.24 | 3.87 |
| WhatsHap | 0.69 | 2.78 | 6.51 | 11.32 | 0.64 | 2.44 | 12.09 | 12.77 | 1.41 | 3.27 | 7.88 | 13.86 |
| Ralphi | 1.13 | 2.83 | 5.27 | 15.43 | 0.97 | 1.66 | 2.73 | 12.58 | 1.86 | 3.18 | 4.39 | 7.18 |
| Memory (GB) | 1.40 | 1.99 | 3.54 | 11.50 | 1.40 | 1.61 | 2.57 | 12.22 | 1.20 | 1.79 | 2.38 | 4.22 |
[i] Memory usage is reported for Ralphi. Runtime is an average over three separate runs.
Discussion
In this work, we motivate the use of DRL for read-based haplotype assembly using the reduction of this task to the NP-hard MFC objective on fragment graphs. Our approach leverages (1) GNNs to learn effective graph state representations for complex and heterogeneous fragment graph topologies and (2) an RL model to learn the optimal policy for partitioning the fragments into haplotypes while maximizing the MFC objective. Our results show that this approach can learn an effective strategy for haplotype assembly and outperforms the state of the art using inputs from different sequencing platforms. We expect its performance to improve as we compile larger and more diverse training data sets and further optimize the framework. It is important to note, however, that the accuracy of the assembled haplotypes also significantly depends on the accuracy of the input alignments and variant calls; because read alignment and variant calling errors are more prevalent in highly repetitive regions of the genome, these regions still remain challenging to phase. As future work, we plan to train and evaluate models using inputs from additional sequencing technologies (e.g., Hi-C) and cross-technology combinations, extend this work to phase indels and SVs, and investigate additional strategies to address the errors and artifacts inherited from upstream data analysis steps. In addition to haplotype assembly, we hope that this work can also motivate the development of other DRL-based tools for key problems in genomics that have been reduced to NP-hard formulations and that are typically solved using expert-driven heuristics.
Methods
Fragment graph construction
Allele matrix
Given a set of read alignments and heterozygous variants, the input to a read-based phasing algorithm is usually represented as a matrix M of size m × n, where m is the number of fragments (informative read alignments that span at least two heterozygous variants), n is the number of heterozygous variants (e.g., SNPs), and each entry, Mi,j, stores the allele of fragment Fi covering the locus of variant j, with the reference allele usually encoded as zero and the variant allele as one, and − is used when the fragment does not cover the variant.
Fragment graph
As described previously (Duitama et al. 2010), the fragment graph, G = (V, E) is constructed from an allele matrix M, such that V = {1, …, m} and (u, v) ∈ E if and only if w(u, v) ≠ 0, where and o(a, b) is defined as follows. Given two alleles a and b,
As such, each node in a fragment graph corresponds to a fragment Fi in matrix M, and an edge connects two nodes only if the two corresponding fragments cover at least one common variant. The weight of each edge represents the difference in the number of alleles that the two fragments agree on and disagree on, respectively, such that stronger agreement will result in a negative edge weight, and a stronger conflict will result in a positive edge weight. A cut in this graph naturally represents an assignment of fragments to each haplotype. Intuitively, higher-weight cuts will separate fragments with a higher number of conflicting alleles, which are likely to pertain to different haplotypes.
Maximum fragment cut
The MFC optimization model proposed by Duitama et al. (2010) solves the read-based haplotype assembly problem by finding the maximum weighted cut in the fragment graph G. Namely, let C be a subset of nodes representing a cut of G; the fragment cut score is then defined as . The MFC objective is to find the cut that maximizes s(C).
Note that fragment graphs are not usually connected, and in practice, MFC is computed for each connected component of the graph separately, resulting in an output that typically consists of multiple distinct phased haplotype sets. The accuracy and length of the resulting partial haplotypes in each set are key metrics used to evaluate haplotype assembly as described below.
Fragment graph compression
To scale to higher coverage data sets, Ralphi adds an additional graph compression step during construction. In particular, we combine all equivalent fragments into a single node in G with an associated compression number c, in which fragments are considered equivalent if they cover the same set of variants with the same alleles (note this can also be extended to include fully complementary fragments, which have different alleles at all variant sites). As a result, edges between equivalent fragments are removed (these are by definition edges that capture perfect fragment agreement and will not be split by an optimal cut), and the weight w(u, v) of the remaining edges is multiplied by c(u) * c(v), corresponding to the compression numbers of the two connected nodes, respectively. By construction, this compression procedure guarantees that the MFCs of the compressed and original graphs are equivalent.
Preprocessing: read and variant selection
Similar to other read-based phasing tools, Ralphi performs an initial selection of informative read alignments (provided as an input BAM file) and variants (provided as a VCF file). We primarily rely on previously established best quality control practices for this preprocessing step. For example, Ralphi performs alignment filtration based on the alignment MAPQ score, as well as read quality. To improve the accuracy of alleles observed at variant sites in long-read sequencing, which have a higher occurrence of indel errors, Ralphi calls into the realignment module of WhatsHap, which realigns a small window of the read around the variant site. To increase fragment contiguity, Ralphi joins nonoverlapping partial alignments of the same read into a single fragment. Finally, given the selected reads, Ralphi examines the evidence at each variant site and removes variants that are covered only by a single SNP allele, by a single SNP allele and an indel, or only by reads from a single strand (similar to LongPhase) (Lin et al. 2022).
DRL framework for MFC
Fragment graph embedding
Background
GNNs are a powerful paradigm for graph representation learning and can effectively capture complex combinatorial graph structures (Xu et al. 2019). Briefly, GNNs rely on a message-passing scheme, wherein each node aggregates features from its neighbors to update its representation. Starting from an initial set of node features, the representation of each node is updated iteratively using message passing (with the number of iterations given by the number of network layers), resulting in a final z-dimensional embedding vector.
GNN architecture
Ralphi uses a GCN (Kipf and Welling 2017) to embed fragment graphs. Our network consists of a single GCN layer with the ELU activation function (Clevert et al. 2015) and z = 264.
Input node features
Because the embedding is used to capture the state of the fragment graph after each iteration (i.e., after the assignment of a single node to one of the two haplotypes), each node in the graph is associated with a feature representing its current haplotype assignment. A node can be in three possible states: unassigned, assigned to haplotype 1 (H1), or assigned to haplotype 2 (H2). Additionally, Ralphi includes betweenness centrality as a node feature, which encodes the fraction of shortest paths passing through a given node w and is defined as , where σ(u, v) denotes the number of shortest paths between u and v, whereas σ(u, v|w) is the number of shortest paths from u to v that pass through w. Because computing all shortest paths takes Θ (|V|3) time, we use the approximate estimation of betweenness centrality proposed by Brandes and Pich (2007) applied to the unweighted fragment graph.
RL architecture
Background
Briefly, in DRL, we define a set of actions A, states S, and a reward function R(s, a) that specifies the amount of reward received by the agent when taking an action a from state s; we then train an agent to learn a policy that maximizes its reward through trial and error.
States, actions, and reward
Based on the max-cut formulation by Khalil et al. (2017), (1) Ralphi states correspond to the fragment graph with partial haplotype assignments represented by the graph embeddings; (2) actions involve selecting an unassigned node in the graph and assigning it to one of the two haplotypes; and (3) the reward is given by the change in fragment cut value when assigning a node u to haplotype, Hk, namely, , where Nbrs(u) represents all the neighbors of node u in the graph, which have already been assigned to a haplotype.
RL algorithm
Ralphi uses the popular actor-critic RL method (Konda and Tsitsiklis 1999) to train the agent. This method consists of two networks, the actor and the critic, such that the actor learns which actions the agent should take and the critic learns the value of different actions. In Ralphi, the actor and the critic are single linear layer networks, with weights initialized using an orthogonal matrix to mitigate the vanishing gradient phenomenon; each layer is also normalized using spectral normalization, which has been shown to increase actor-critic's stability (Cetin et al. 2022).
Training data generation
To generate synthetic read data sets, we selected 10 genomes from the 1000 Genomes Project for model training from different populations and used BCFtools (Danecek et al. 2021) to incorporate the variants from each genome into the GRCh38 reference. We then generated synthetic Illumina and ONT reads from each genome. For short reads, we used the DWGSIM read simulator (https://github.com/nh13/DWGSIM/) with 2% and 5% error rates and aligned reads with BWA-MEM (Li 2013). For ONT reads, we used PBSIM3 (Ono et al. 2022) for read simulation and minimap2 (Li 2018) for read mapping (using the ONT-specific preset). We used SAMtools downsampling (Danecek et al. 2021) to produce synthetic data sets at 5×, 10×, 15×, and 30× coverage. Similarly, we also downsampled real read data sets to produce graphs for 5×, 10×, 15×, and 30× coverage regimes. We removed fragment graphs larger than 5000 nodes to decrease the training time. We used curriculum learning (Bengio et al. 2009) during training, which shows progressively harder examples to the agent as it trains, by ordering our graphs from the smallest and sparsest to the largest and most dense. Fragment graphs generated from Chromosome 1 and Chromosome 20 were held out for testing. Supplemental Notes 1 and 2 provide the specific commands and parameters used to obtain the read data sets and fragment graphs used for model training, as well as the training parameters.
Evaluation metrics
We used the following standard metrics to evaluate phasing results: switch error rate (Duitama et al. 2012), mismatch error rate (Edge et al. 2017), and the AN50 score (Duitama et al. 2012). The switch and mismatch error rates evaluate the accuracy of the haplotype assignments, whereas the AN50 score evaluates the contiguity of haplotype blocks. A switch error occurs when a pair of adjacent variants are incorrectly phased compared with the ground-truth haplotypes. If two switch errors occur in a row at consecutive positions, they are counted as a mismatch error instead (also referred to as a short switch error). The switch error rate and the mismatch error rate are obtained by dividing the number of respective errors by the number of positions at which they can occur, respectively. The AN50 metric is the adjusted N50 metric, where the N50 represents the maximal span in base pairs such that half of all phased variants are in a block larger than the span. To account for unphased variants, the AN50 metric adjusts the N50 by multiplying the span of a block by the fraction of phased variants it covers. We used the utility script provided by HapCUT2 to compute these metrics.
Training and evaluation data sources
The two NA12878 ONT R10 data sets were downloaded from (1) s3://ont-open-data/giab_2023.05/analysis/variant_calling/hg001_sup_all/hg001.haplotagged.bam and (2) https://gtgseq.s3.amazonaws.com/ont-r10-dna/NA12878/analyses/basecalls/guppy642hac/PGXXHX230142_guppy642hac_mm217.bam. The HG002 ONT data set is available at gs://fc-46bf051e-aec3-4adb-8178-3c51bc5e64ae/HG002_R10/reads/GM24385_R10_638.bam. The NA12878 Illumina reads were downloaded from ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR323/ERR3239334/NA12878.final.cram. The HG002 Illumina reads were downloaded from Genome in a Bottle at https://ftp.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/HG002.GRCh38.2x250.bam. The PacBio HiFi reads were downloaded from https://www.ncbi.nlm.nih.gov/bioproject/PRJNA540705 (NA12878) and https://downloads.pacbcloud.com/public/dataset/HG002-CpG-methylation-202202/HG002.GRCh38.haplotagged.bam (HG002). The 1000 Genomes Project variants were downloaded from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20220422_3202_phased_SNV_INDEL_SV/. The NA12878 truth set is available on the Illumina Platinum Genomes GitHub (https://github.com/Illumina/PlatinumGenomes). The HG002 truth set is available on the Genome in a Bottle platform at https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio/HG002_NA24385_son/NISTv4.2.1/GRCh38/SupplementaryFiles/HG002_GRCh38_1_22_v4.2.1_benchmark_hifiasm_v11_phasetransfer.vcf.gz. The real short-read data sets from the 1000 Genomes Project (for the 10 genomes used in model training) were downloaded from ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3988804/HG00627.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3240148/HG00142.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3240178/HG00234.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3242450/HG03046.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3242065/HG01598.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3241781/HG00250.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3242545/HG03388.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3243142/HG04225.final.cram, ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR324/ERR3242343/HG02047.final.cram, and ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR323/ERR3239643/NA11932.final.cram. The ONT R10 data sets for six additional genomes used in training were obtained from https://s3.amazonaws.com/1000g-ont/index.html?prefix=ALIGNMENT_AND_ASSEMBLY_DATA/100_PLUS/IN-HOUSE_MINIMAP2/.
Software availability
The Ralphi code, models, and documentation are available on GitHub (https://github.com/PopicLab/ralphi) under the BSD-3 license and as Supplemental Code.
Competing interest statement
V.P. owns shares of Illumina. The remaining authors have no competing interests to declare.
Acknowledgments
This work was supported by the Broad Institute Schmidt Fellowship (V.P.), National Institutes of Health (NIH) R21HG013567 (V.P.), and NIH 1R35GM141861 (B.B.). We thank the authors of WhatsHap for providing a robust and well-documented software framework, which allowed us to easily use and extend its read selection and realignment modules for preprocessing.
Author contributions: V.P. conceived and designed the study. V.P., E.B., and A.M. developed the software and performed analyses. E.B. and A.M. benchmarked existing methods with contributions from B.E. and generated large-scale training data sets. E.B., A.M., B.B., and V.P. performed graph feature analysis. E.B. performed model and parameter tuning. V.P. wrote the manuscript, with contributions from B.B., E.B., and A.M. V.P. supervised the research. All authors reviewed the manuscript.
Notes
[2] Supplementary material [Supplemental material is available for this article.]
[3] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280569.125.
References
- ↵The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393
- ↵Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500: 207–211. 10.1038/nature12064
- ↵Aguiar D, Istrail S. 2012. HapCompass: a fast cycle basis algorithm for accurate haplotype assembly of sequence data. J Comput Biol 19: 577–590. 10.1089/cmb.2012.0084
- ↵Bansal V, Bafna V. 2008. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics 24: i153–i159. 10.1093/bioinformatics/btn298
- ↵Bengio Y, Louradour J, Collobert R, Weston J. 2009. Curriculum learning. In ICML '09: The 26th Annual International Conference on Machine Learning held in conjunction with the 2007 International Conference on Inductive Logic Programming, Montreal, pp. 41–48. Association for Computing Machinery, New York.
- ↵Beretta S, Patterson MD, Zaccaria S, Della Vedova G, Bonizzoni P. 2018. HapCHAT: adaptive haplotype assembly for efficiently leveraging high coverage in long reads. BMC Bioinformatics 19: 252. 10.1186/s12859-018-2253-8
- ↵Berger E, Yorukoglu D, Peng J, Berger B. 2014. HapTree: a novel Bayesian framework for single individual polyplotyping using NGS data. PLoS Comput Biol 10: e1003502. 10.1371/journal.pcbi.1003502
- ↵Berger E, Yorukoglu D, Zhang L, Nyquist SK, Shalek AK, Kellis M, Numanagić I, Berger B. 2020. Improved haplotype inference by exploiting long-range linking and allelic imbalance in RNA-seq datasets. Nat Commun 11: 4662. 10.1038/s41467-020-18320-z
- ↵Bonizzoni P, Dondi R, Klau GW, Pirola Y, Pisanti N, Zaccaria S. 2016. On the minimum error correction problem for haplotype assembly in diploid and polyploid genomes. J Comput Biol 23: 718–736. 10.1089/cmb.2015.0220
- ↵Brandes U, Pich C. 2007. Centrality estimation in large networks. Int J Bifurcation Chaos 17: 2303–2318. 10.1142/S0218127407018403
- ↵Brinton J, Ramirez-Gonzalez RH, Simmonds J, Wingen L, Orford S, Griffiths S, Project WG, Haberer G, Spannagl M, Walkowiak S, 2020. A haplotype-led approach to increase the precision of wheat breeding. Commun Biol 3: 712. 10.1038/s42003-020-01413-2
- ↵Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. 10.1086/521987
- ↵Browning SR, Browning BL. 2011. Haplotype phasing: existing methods and new developments. Nat Rev Genet 12: 703–714. 10.1038/nrg3054
- ↵Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562: 203–209. 10.1038/s41586-018-0579-z
- ↵Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, 2022. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185: 3426–3440.e19. 10.1016/j.cell.2022.08.004
- ↵Cetin E, Chamberlain B, Bronstein M, Hunt JJ. 2022. Hyperbolic deep reinforcement learning. arXiv:2210.01542 [cs.LG]. 10.48550/arXiv.2210.01542
- ↵Cilibrasi R, Van Iersel L, Kelk S, Tromp J. 2007. The complexity of the single individual SNP haplotyping problem. Algorithmica 49: 13–36. 10.1007/s00453-007-0029-z
- ↵Clevert DA, Unterthiner T, Hochreiter S. 2015. Fast and accurate deep network learning by exponential linear units (ELUs). arXiv:1511.07289 [cs.LG]. 10.48550/arXiv.1511.07289
- ↵Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, 2021. Twelve years of SAMtools and BCFtools. GigaScience 10: giab008. 10.1093/gigascience/giab008
- ↵Delaneau O, Zagury JF, Robinson MR, Marchini JL, Dermitzakis ET. 2019. Accurate, scalable and integrative haplotype estimation. Nat Commun 10: 5436. 10.1038/s41467-019-13225-y
- ↵De Rosa M, Fasano C, Panariello L, Scarano M, Belli G, Iannelli A, Ciciliano F, Izzo P. 2000. Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene 19: 1719–1723. 10.1038/sj.onc.1203447
- ↵Dörk T, Bendix-Waltes R, Wegner RD, Stumm M. 2004. Slow progression of ataxia-telangiectasia with double missense and in frame splice mutations. Am J Med Genet A 126A: 272–277. 10.1002/ajmg.a.20601
- ↵Douglas JA, Boehnke M, Gillanders E, Trent JM, Gruber SB. 2001. Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat Genet 28: 361–364. 10.1038/ng582
- ↵Duitama J, Huebsch T, McEwen G, Suk EK, Hoehe MR. 2010. ReFHap: a reliable and fast algorithm for single individual haplotyping. In BCB '10: ACM International Conference on Bioinformatics and Computational Biology, Niagara Falls, New York, pp. 160–169. Association for Computing Machinery, New York.
- ↵Duitama J, McEwen GK, Huebsch T, Palczewski S, Schulz S, Verstrepen K, Suk EK, Hoehe MR. 2012. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of single individual haplotyping techniques. Nucleic Acids Res 40: 2041–2053. 10.1093/nar/gkr1042
- ↵Eberle MA, Fritzilas E, Krusche P, Källberg M, Moore BL, Bekritsky MA, Iqbal Z, Chuang H-Y, Humphray SJ, Halpern AL, 2017. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 27: 157–164. 10.1101/gr.210500.116
- ↵Edge P, Bafna V, Bansal V. 2017. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27: 801–812. 10.1101/gr.213462.116
- ↵Gamaarachchi H, Samarakoon H, Jenner SP, Ferguson JM, Amos TG, Hammond JM, Saadat H, Smith MA, Parameswaran S, Deveson IW. 2022. Fast nanopore sequencing data analysis with slow5. Nat Biotechnol 40: 1026–1029. 10.1038/s41587-021-01147-4
- ↵Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MHY, 2010. A draft sequence of the neandertal genome. Science 328: 710–722. 10.1126/science.1188021
- ↵Jarvis ED, Formenti G, Rhie A, Guarracino A, Yang C, Wood J, Tracey A, Thibaud-Nissen F, Vollger MR, Porubsky D, 2022. Semi-automated assembly of high-quality diploid human reference genomes. Nature 611: 519–531. 10.1038/s41586-022-05325-5
- ↵Ke Z, Vikalo H. 2020. A graph auto-encoder for haplotype assembly and viral quasispecies reconstruction. In The Thirty-Fourth AAAI Conference on Artificial Intelligence, New York, Vol. 34, pp. 719–726. AAAI Press, Palo Alto, CA.
- ↵Ke Z, Vikalo H. 2022. Deep learning for assembly of haplotypes and viral quasispecies from short and long sequencing reads. In BCB '22: 13th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Northbrook, IL, pp. 1–10. Association for Computing Machinery, New York.
- ↵Khalil E, Dai H, Zhang Y, Dilkina B, Song L. 2017. Learning combinatorial optimization algorithms over graphs. In Advances in Neural Information Processing Systems 30 (NIPS 2017) (ed. Guyon I, et al.), pp. 6348–6358. Curran Associates Inc., Red Hook, NY.
- ↵Kipf TN, Welling M. 2017. Semi-supervised classification with graph convolutional networks. In 5th International Conference on Learning Representations, ICLR 2017, Toulon, France. OpenReview.net. https://openreview.net/forum?id=SJU4ayYgl
- ↵Kolmogorov M, Billingsley KJ, Mastoras M, Meredith M, Monlong J, Lorig-Roach R, Asri M, Alvarez Jerez P, Malik L, Dewan R, 2023. Scalable nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat Methods 20: 1483–1492. 10.1038/s41592-023-01993-x
- ↵Konda VR, Tsitsiklis JN. 1999. Actor-critic algorithms. In Advances in Neural Information Processing Systems 12 (NIPS 1999) (ed. Solla S, et al.), pp. 1008–1014. The MIT Press, Cambridge, MA.
- ↵Kuleshov V. 2014. Probabilistic single-individual haplotyping. Bioinformatics 30: i379–i385. 10.1093/bioinformatics/btu484
- ↵Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. 10.1371/journal.pbio.0050254
- ↵Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]. 10.48550/arXiv.1303.3997
- ↵Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- ↵Lin JH, Chen LC, Yu SC, Huang YT. 2022. LongPhase: an ultra-fast chromosome-scale phasing algorithm for small and large variants. Bioinformatics 38: 1816–1822. 10.1093/bioinformatics/btac058
- ↵Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, Schoenherr S, Forer L, McCarthy S, Abecasis GR, 2016. Reference-based phasing using the haplotype reference consortium panel. Nat Genet 48: 1443–1448. 10.1038/ng.3679
- ↵Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, 2010. Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362: 1181–1191. 10.1056/NEJMoa0908094
- ↵Martin M, Patterson M, Garg S, O Fischer S, Pisanti N, Klau GW, Schöenhuth A, Marschall T. 2016. WhatsHap: fast and accurate read-based phasing. bioRxiv 10.1101/085050
- ↵Miller DB, Piccolo SR. 2021. A survey of compound heterozygous variants in pediatric cancers and structural birth defects. Front Genet 12: 640242. 10.3389/fgene.2021.640242
- ↵Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987
- ↵Ono Y, Hamada M, Asai K. 2022. PBSIM3: a simulator for all types of PacBio and ONT long reads. NAR Genom Bioinform 4: lqac092. 10.1093/nargab/lqac092
- ↵Panconesi A, Sozio M. 2004. Fast hare: a fast heuristic for single individual SNP haplotype reconstruction. In Algorithms in Bioinformatics: 4th International Workshop, WABI 2004, Bergen, Norway, September 17–21, 2004, Proceedings 4, pp. 266–277. Springer, Berlin, Heidelberg.
- ↵Piane M, Molinaro A, Soresina A, Costa S, Maffeis M, Germani A, Pinelli L, Meschini R, Plebani A, Chessa L, 2016. Novel compound heterozygous mutations in a child with ataxia-telangiectasia showing unrelated cerebellar disorders. J Neurol Sci 371: 48–53. 10.1016/j.jns.2016.10.014
- ↵Rahner N, Höefler G, Högenauer C, Lackner C, Steinke V, Sengteller M, Friedl W, Aretz S, Propping P, Mangold E, 2008. Compound heterozygosity for two MSH6 mutations in a patient with early onset colorectal cancer, vitiligo and systemic lupus erythematosus. Am J Med Genet A 146A: 1314–1319. 10.1002/ajmg.a.32210
- ↵Rizzi R, Bafna V, Istrail S, Lancia G. 2002. Practical algorithms and fixed-parameter tractability for the single individual snp haplotyping problem. In Algorithms in Bioinformatics: Second International Workshop, WABI 2002, Rome, Italy, September 17–21, 2002, Proceedings 2, pp. 29–43. Springer, Berlin, Heidelberg.
- ↵Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ. 2011. The importance of phase information for human genomics. Nat Rev Genet 12: 215–223. 10.1038/nrg2950
- ↵Vollger MR, Dishuck PC, Harvey WT, DeWitt WS, Guitart X, Goldberg ME, Rozanski AN, Lucas J, Asri M, 2023. Increased mutation and gene conversion within human segmental duplications. Nature 617: 325–334. 10.1038/s41586-023-05895-y
- ↵Welch KO, Marin RS, Pandya A, Arnos KS. 2007. Compound heterozygosity for dominant and recessive GJB2 mutations: effect on phenotype and review of the literature. Am J Med Genet A 143A: 1567–1573. 10.1002/ajmg.a.31701
- ↵Xu K, Jegelka S, Hu W, Leskovec J. 2019. How powerful are graph neural networks? In 7th International Conference on Learning Representations, ICLR 2019, New Orleans.
- ↵Xue H, Rajan V, Lin Y. 2022. Graph coloring via neural networks for haplotype assembly and viral quasispecies reconstruction. In Proceedings of the 36th International Conference on Neural Information Processing Systems. Curran Associates Inc., Red Hook, NY.
- ↵Yu Y, Chen L, Miao X, Li SC. 2021. SpecHap: a diploid phasing algorithm based on spectral graph theory. Nucleic Acids Res 49: e114. 10.1093/nar/gkab709
- ↵Zook JM, Hansen NF, Olson ND, Chapman L, Mullikin JC, Xiao C, Sherry S, Koren S, Phillippy AM, Boutros PC, 2020. A robust benchmark for detection of germline large deletions and insertions. Nat Biotechnol 38: 1347–1355. 10.1038/s41587-020-0538-8