Unlike most Ascomycete fungi, which live primarily as plant pathogens or plant saprobes, Coccidioides is capable of causing life-threatening disease in immunocompetent mammals, including humans. As the causal agent of coccidioidomycosis or “Valley fever,” Coccidioides infects at least 150,000 people annually, ∼40% of whom develop a pulmonary infection (Hector and Laniado-Laborin 2005). However, a chronic and disseminated form of coccidioidomycosis, for which existing treatments can be prolonged and difficult to tolerate, occurs in roughly 5% of patients (Galgiani et al. 2005). The virulent nature of this fungus and its potential for dispersal by airborne spores led to its listing as a U.S Health and Human Services Select Agent (Dixon 2001) and has fueled efforts to develop an effective vaccine and new treatments (Hung et al. 2002; Hector and Rutherford 2007).
Coccidioides is an environmentally acquired, dimorphic pathogen. When not infecting a mammal, the fungus lives in the arid, alkaline New World deserts, where it is believed to grow as a filamentous soil saprophyte (Papagiannis 1967; Fisher et al. 2007). The filaments produce asexual spores (arthroconidia), which are inhaled to initiate infection. Once in the lungs, arthroconidia enlarge into spherules, documenting a morphological switch from polar to isotrophic growth. Spherules subsequently differentiate to produce internal spores (endospores) that are released upon spherule rupture. This latter morphology, endospore-containing spherules, is unique to Coccidioides among all known Ascomycota. Endospores are capable of disseminating in the host and re-initiating the spherulation cycle, but the host can sequester spherules in a granuloma to prevent disease dissemination. In the absence of a successful host response, chronic infection may persist for at least 12 yr (Hernandez et al. 1997), although human disease can progress to death in a much shorter period. Upon host death, the fungus reverts to filamentous growth and the production of arthroconidia.
Coccidioides is composed of two closely related species, Coccidioides immitis and Coccidioides posadasii, and is a member of the Onygenales, an order characterized by many species that also tend to associate with animals. However, despite this shared characteristic, recently diverged fungal relatives of Coccidioides, such as Uncinocarpus, Gymnoascus, and Crysosporium species, are not known to cause disease (Untereiner et al. 2004). This observation has led to the parsimonious theory that Coccidioides recently acquired its pathogenic phenotype. Although a great deal is known about the clinical aspects of coccidioidomycosis and the biology of this fungus in laboratory mice, relatively little is known about the life history of Coccidioides between infections (Barker et al. 2007) or how it evolved the ability to cause disease in immunocompetent mammals. To address these questions, the genomes of C. immitis and C. posadasii, as well as their Onygenlean relatives Uncinocarpus reesii, a nonpathogenic fungus, and Histoplasma capsulatum, a mammalian pathogen, were sequenced and compared with those of 13 more distantly related Ascomycota, 12 of which associate with plants. Comparing the Coccidioides genome sequences across a range of evolutionary distances resolved various levels of genome evolution, including changes in gene family size, gene gain and loss, and the detection of positive natural selection, and provided an evolutionary context for observed differences between the taxa. Ultimately, the adoption of such a hierarchical comparative genomics approach reveals that myriad genomic changes are involved in shaping the evolution of phenotype and, in the case of Coccidioides, elucidates an understanding of how the fungus evolved to associate with animals.
Results
Genome sequencing and annotation
The C. posadasii C735 genome was sequenced at 8× coverage and assembled into 58 contigs that totaled 27 Mb. Sequenced at 14× coverage, the C. immitis RS genome assembled into seven contigs and totaled 28.9 Mb. While similar in size, these genomes differ in the number of annotated genes, with 10,355 in C. immitis and 7229 in C. posadasii (Table 1). This variation most likely results from the use of different annotation methodologies by the sequencing institutions (see Methods). In particular, gene splitting and fusion occurred during annotation, as evidenced by the 9996 C. immitis genes that have BLASTN hits with greater than 90% identity in the C. posadasii genome. The comparative analyses presented here employed a conservative approach, considering only those genes annotated in both species.
Although the nonrepetitive sequence of these genomes differs only by 400 kb, there is a large difference in repetitive DNA (C. immitis 17%, C. posadasii 12%) that accounts for an additional 1.84 Mb of long, interspersed, repetitive sequence in C. immitis (Table 2). U. reesii and H. capsulatum also have repeated regions (4% and 19%, respectively), but neither has a bias toward high copy number repeats (medium or distributed across low, medium, and high, respectively).
Distribution of repetitive genomic content
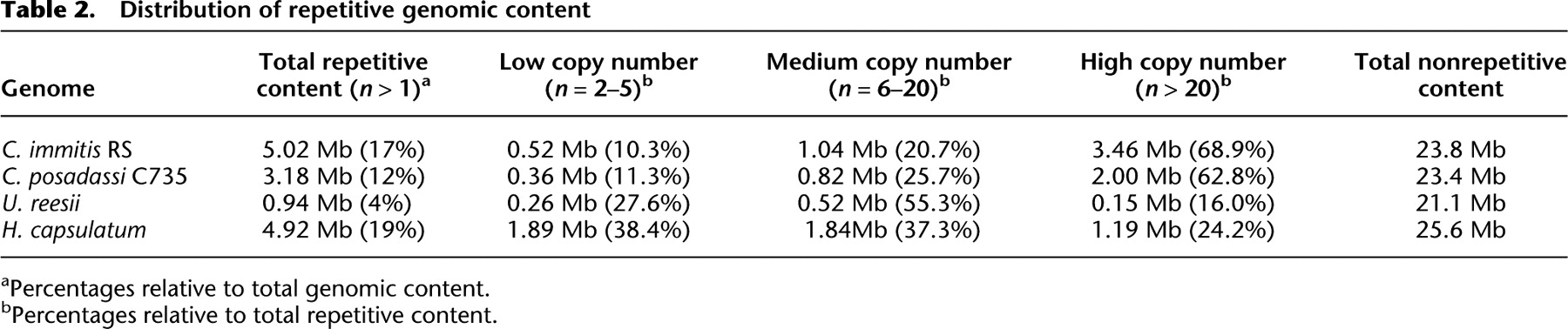
[i] aPercentages relative to total genomic content.
[ii] bPercentages relative to total repetitive content.
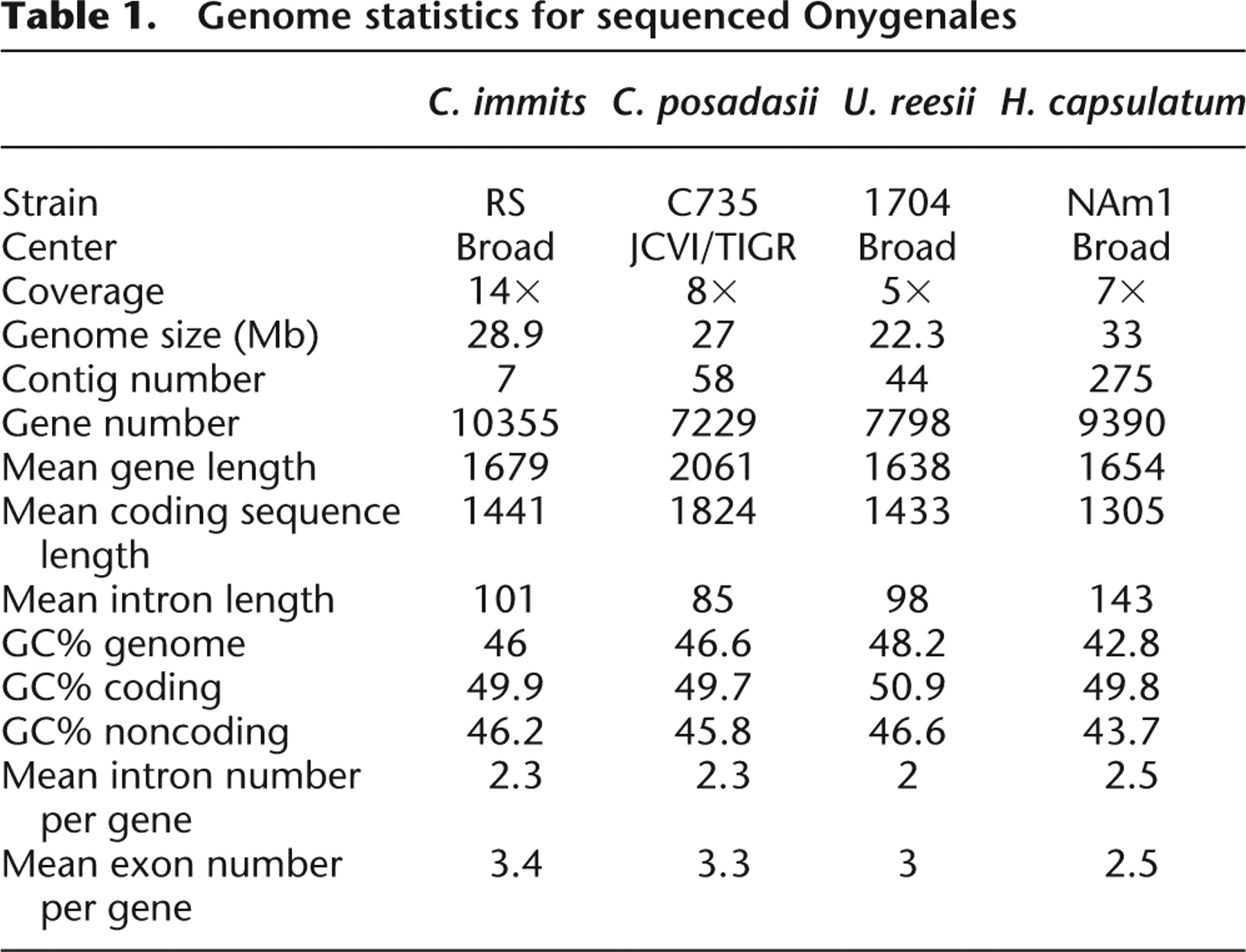
In the Coccidioides genomes, the GC content of repeats is 14%–15% lower than the GC content of nonrepetitive sequence (Fig. 1). Furthermore, in C. immitis, CpG dinucleotides are 19 times more abundant in nonrepetitive sequence than in repetitive sequence. In contrast to an average CpG frequency of 51 per kilobase in nonrepetitive sequence, 73% of the repetitive regions have no CpG dinucleotides and there are contiguous stretches of 100-kb windows with a CpG frequency as low as 0.08 per kilobase. No other dinucleotide exhibits such a dramatic bias across repeats.
Evidence of mutational bias associated with repetitive regions of the Coccidioides genomes. For both the C. immitis and C. posadasii genomes, each 1-kb nonoverlapping window of sequence was categorized into a repeat class. Classification is based on the number of homologous copies (n) of that window identified within the genome: nonrepetitive, low (n < 6), mid (n = 6–20), and high (n > 20). The mean GC percentage, the mean CpG frequency, and the fraction of elements with a CpG frequency less than 0.010 were measured for each repeat class. The results indicate that repeats in Coccidioides tend to exhibit a paucity of CpG dinucleotides compared with nonrepetitive regions.
Coccidioides chromosome structure and conserved synteny
Coccidioides species are estimated to have four chromosomes by CHEF gel analysis (Pan and Cole 1992). This estimate may be low because supercontigs three and four in the high-quality C. immitis genome sequence assembly are similar in size at 4.66 Mb and 4.32 Mb, respectively. Five distinct chromosomes, which accommodate 29 contigs, are seen in an optical map of the C. posadasii genome and support a higher estimate (Supplemental Fig. 1). The C. immitis genome has seven supercontigs, but a whole-genome sequence alignment between C. immitis and C. posadasii supports an estimate of five chromosomes (Supplemental Fig. 2). Two C. immitis supercontigs (five and six) are linked to the fifth chromosome of C. posadasii. The extremely short C. immitis seventh contig (0.47 Mb) exhibits no unique, colinear homology with C. posadasii and likely represents an insertion in C. immitis.
Species-specific differences in chromosome structure were identified where <500 bp of homologous sequence was found when comparing 1-kb windows across the C. immitis and C. posadasii genomes. Of the 23.8 Mb of nonrepetitive sequence in C. immitis, 22.3 Mb (93.5%) exhibits homology with the C. posadasii genome, with a median sequence identity of 98.3%. Thus, 1.5 Mb of nonrepetitive C. immitis DNA lacks significant similarity to C. posadasii. The reciprocal analysis confirms the 22.3 Mb of homologous sequence and identifies 1.1 Mb of nonrepetitive C. posadasii DNA that is absent from C. immitis. A parallel analysis using only the annotated protein coding genes from these two genomes identifies 282 C. immitis-specific genes and 66 C. posadasii-specific genes (Supplemental Table 3).
Intersecting the window- and gene-based analyses identifies 53 species-specific, gene-containing regions in C. immitis and 45 such regions in C. posadasii (Supplemental Table 4). Because 37 of the 53 C. immitis regions correspond to a location in the C. posadasii genome that is completely bounded within a sequence contig, it is unlikely that these regions are the result of assembly artifacts. In addition, expressed sequence tag (EST) data from the two species provide expression evidence for 132 C. immitis genes and 42 C. posadasii genes in these regions. Interestingly, three of the four regions greater than 100 kb in length are on chromosome 4 and account for 120 of the 282 C. immitis-specific genes. Many of these regions are very close to the ends of the chromosomes, with five of the most proximate regions in C. immitis separated from the chromosomal ends by fewer than five genes, and the genomic locations of six of the C. posadasii-specific regions are syntenically identical to C. immitis-specific regions.
Eurotiomycetes phylogeny and divergence times
Three phylogenetic analyses (distance, likelihood, and Bayesian) were conducted on 2891 aligned orthologs conserved between 16 Eurotiomycetes and the two Sordariomycetes used to root the tree, Sclerotinia sclerotiorum and Fusarium graminearum. Each analysis found the same topology, which is consistent with previous studies (Geiser et al. 2006; James et al. 2006). This well-supported phylogeny (all internal branches had 100% bootstrap support), with median neighbor-joining branch lengths, served as the basis for subsequent genome comparisons (Supplemental Fig. 5).
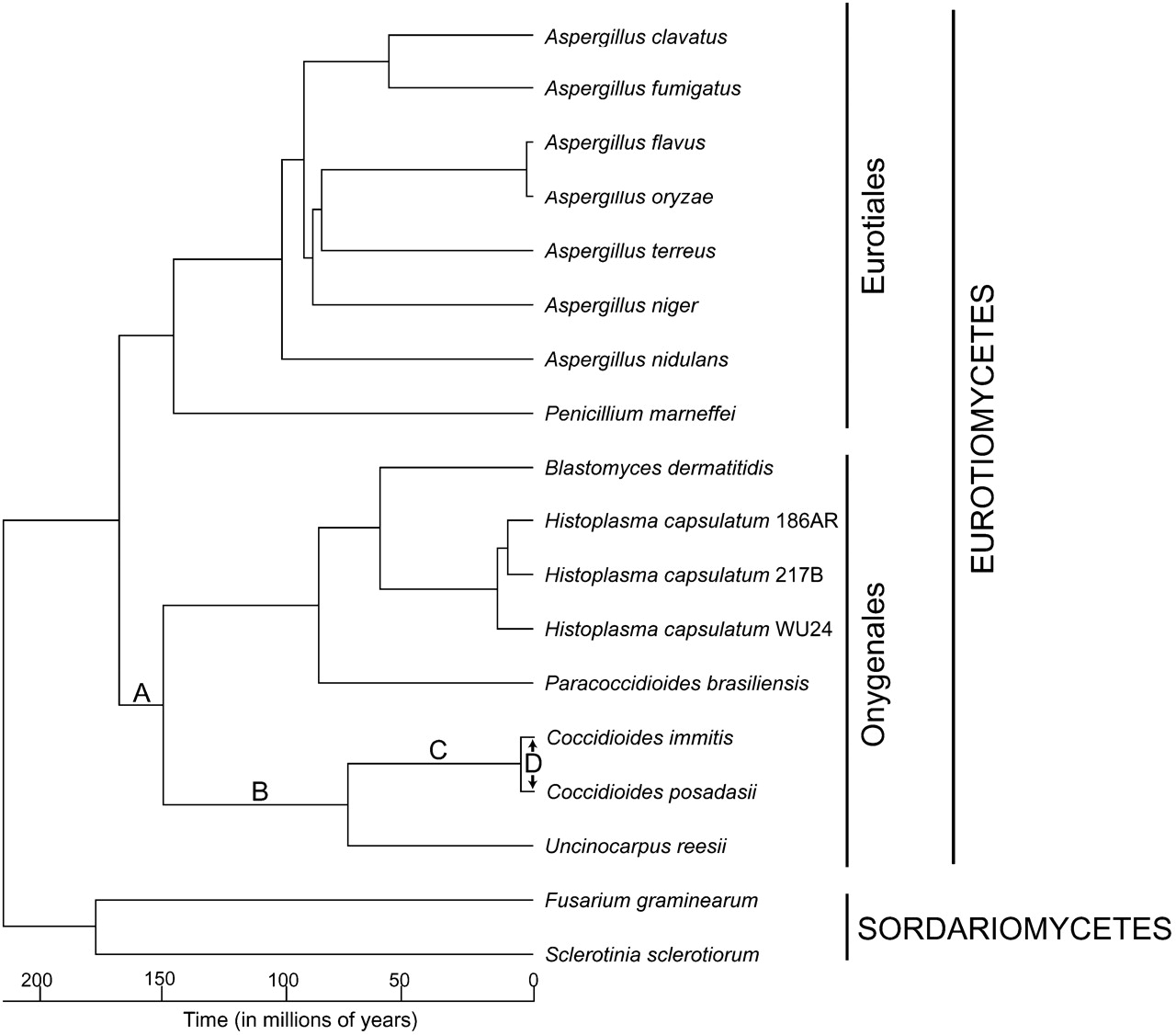
Prior work incorporating fossil data estimated that the Eurotiomycetes and Sordariomycetes diverged roughly 215 million years ago (Mya) (Taylor and Berbee 2006). This calibration point and the NPRS method in the r8s phylogenetic software (Sanderson 2003) enabled divergence time estimation among the Eurotiomycetes represented in the phylogeny (Fig. 2; Supplemental Table 6). The subsequent prediction that C. immitis and C. posadasii diverged 5.1 Mya suggests that these species diverged much more recently than previously estimated (Koufopanou et al. 1998; Fisher et al. 2000).
A timeline of genomic changes that underlie the evolution of pathogenicity in Coccidioides. A RAxML-generated maximum likelihood phylogeny based on an alignment of 1148 concatenated genes that comprise 46,890 unambiguously aligned amino acid sites following the removal of gaps was used as a starting point to perform nonparametric rate smoothing. Divergence times were subsequently estimated from the smoothed phylogeny by applying a calibration of 215 Mya for the Pezizomycotina origin as in the method of Taylor and Berbee (2006). The significant evolutionary events identified in the hierarchical comparative genomic analysis of the Coccidioides lineage are overlaid onto the phylogeny. Taken together, they suggest that a gradual progression of evolutionary events ultimately yielded the pathogenic phenotype of Coccidioides. (A) Gene family reductions associated with the metabolism of plant material were coupled with a growth substrate transition from plant to animal material. (B) Subsequent expansions in proteases and keratinases led to a nutritional association with mammals in a nonpathogenic fashion. (C) The acquisition and adaptation of genes involved in metabolism, membrane biology, and mycotoxin production led to metabolic and morphological phenotypes that enabled survival within a live host, ultimately resulting in disease. (D) Secreted proteins, metabolism genes, and secondary metabolism genes that are shared between the two Coccidioides species have been subject to positive selection since they diverged. This suggests that Coccidioides may experience on-going adaptation in response to host immune system selection pressures.
Gene family expansions and contractions
A phylogenetic analysis conducted on 2798 gene families identified significant changes in gene family size between the Onygenales and their sister order, the Eurotiales, which are primarily associated with plants, as are the outgroup taxa. There are 1043 families that are conserved in size between the orders (Supplemental Table 7). Evaluating those families with size changes with the statistical testing method employed by De Bie et al. (2006) reveals 13 that are significantly smaller among the Onygenales and two that are significantly larger (P < 0.05; Fig. 3).
The evolution of gene family size in the Eurotiomycetes gene family size evolution was evaluated across seven Eurotiomycetes and two Sordariomycete outgroups. The columns capture the size of selected gene families across the taxa, with Coccidioides gene family contractions and expansions represented by numbers in the light and dark gray boxes, respectively (specific information regarding gene family function can be found in the main text). Taxa are listed on the lefthand side according to their phylogenetic orientation and represented by four letter identifiers: Anid indicates Aspergillus nidulans; Afum, Aspergillus fumigatus; Ater, Aspergillus terreus; Hcap, Histoplasma capsulatum; Uree, Uncinocarpus reesii; Cimm, Coccidioides immitis; Cpos, Coccidioides posadasii; Ncra, Neurospora crassa; and Fgra, Fusarium graminearum.
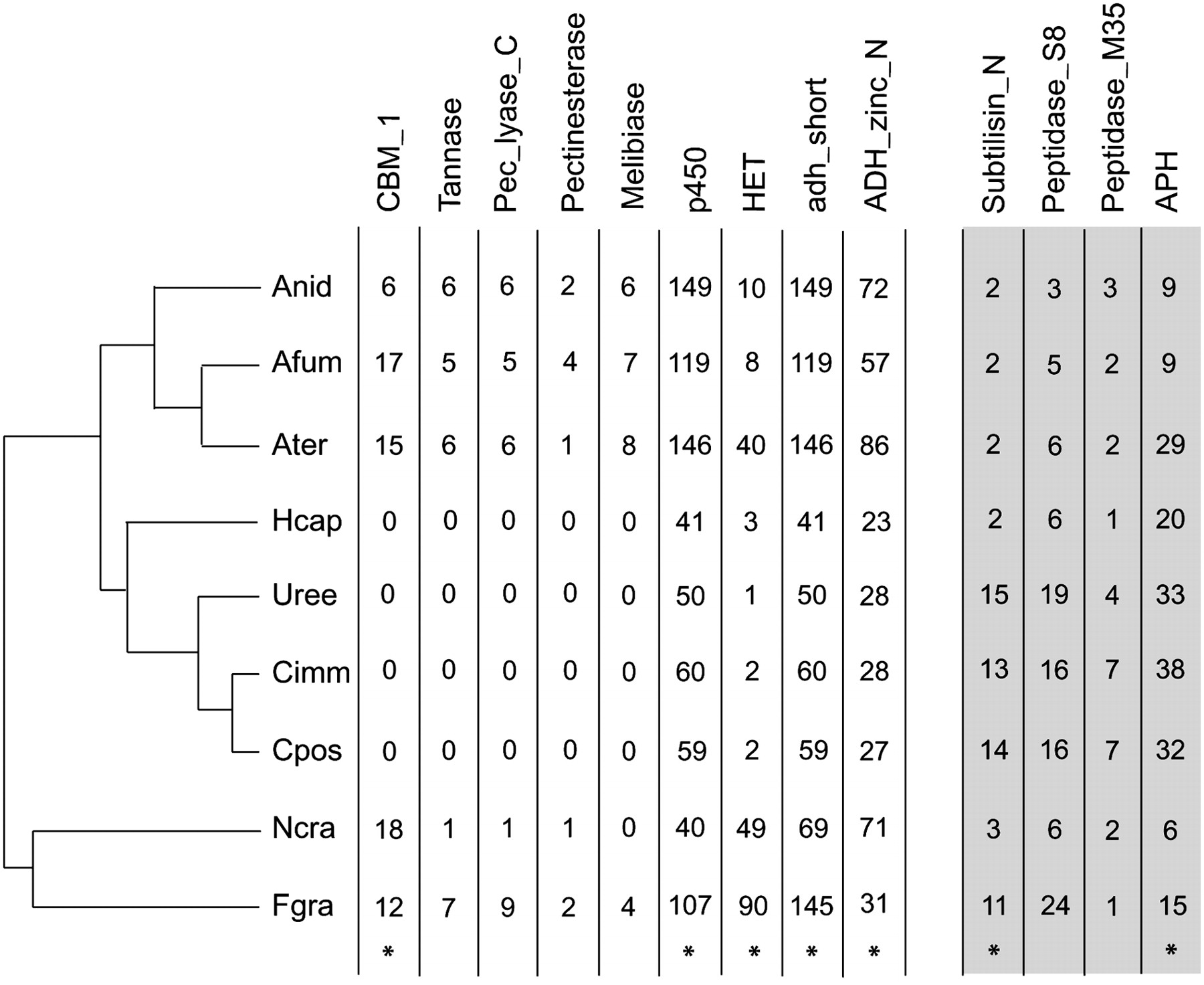
Included in the significantly reduced Onygenales gene families are those that are part of the NADP Rossman clan, including short chain dehydrogenases and zinc-binding dehydrogenases. Additionally, the Onygenales possess relatively few heterokaryon incompatibility (HET) proteins (one to three in Onygenales compared with eight to 40 in Eurotiales), homologs of which have been shown to play a role in prohibiting vegetative fusion between genetically incompatible individuals (Espagne et al. 2002).
Because of the lifestyle differences between the orders, the most interesting significantly reduced gene family in the Onygenales is the fungal cellulose binding domain-containing family. None of the four Onygenales genomes contain copies of this gene, but the rest of the Pezizomycotina evaluated have many. The same pattern of Onygenales lacking genes associated with decay of plants that are found in the Eurotiales and outgroups was also seen for tannase (Onygenales, zero; Euotiales, five to six), cellulase (Onygenales, one to two; Euotiales, 10–13), cutinase (Onygenales, one to two; Euotiales, four to eight) melibiase (Onygenales, zero; Euotiales, six to eight), pectate lyase (Onygenales, zero; Euotiales, four to eight), and pectinesterase (Onygenales, zero; Euotiales, one to four). In these cases, the difference between the Onygenales and Eurotiales was not large enough to be judged statistically significant; however, assuming that the absence of genes is not the result of homology detection sensitivity limitations, the biological significance between any gene and no gene seems unassailable.
Only two families are significantly expanded in the Onygenales. The first, which is expanded in all of the Onygenales genomes evaluated, is the APH phosphotransferase family. In eukaryotes, this family is associated with protein kinase activity, specifically homoserine kinase, fructosamine kinase, and tyrosine kinase activity, while in prokaryotes they are involved in aminoglycoside antibiotic inactivation. The second is the subtilisin N domain-containing family, which is only expanded in Coccidioides and U. reesii. These extracellular serine-proteases are implicated in the pathogenic activity of several fungi (Segers et al. 1999; da Silva et al. 2006), including Aspergillus fumigatus (Monod et al. 2002), and are important virulence factors in many prokaryotes (Henderson and Nataro 2001). Subtilisin N domains often associate with a peptidase S8 family domain, which includes several well-described keratinolytic subtilases (keratinases) (Descamps 2005; Cao et al. 2008). The peptidase S8 family is three times larger in Coccidioides and U. reesii compared with the other taxa, but the difference is not significant. However, the statistical test employed is conservative because it evaluates only the total family size along a lineage and not the phylogenetic information of each member. Evaluating the gene phylogeny of this family reveals that this excess of peptidase S8 members is indeed the result of gene duplications along the lineage shared between Coccidioides and U. reesii (Supplemental Fig. 8). A similar pattern is observed for the deuterolysin metalloprotease (M35) family (Supplemental Fig. 9), which is homologous to MEP1, a known Coccidioides virulence factor (Hung et al. 2005), in that the gene phylogeny reveals duplications along the lineage shared between U. reesii and Coccidioides. However, in this family, Coccidioides has acquired three additional members since it diverged from U. reesii.
Gene gain and loss
Adaptation and changes in virulence can result from lineage-specific gene gain (Censini et al. 1996; Lawrence 2005). The 6250 orthologs shared between U. reesii and at least one Coccidioides species represent the minimum set of core genes present in the ancestor of Coccidioides and its closest relative with a sequenced genome. While the deficit of genes in this ortholog set relative to the total number of Coccidioides genes suggests that lineage-specific gene gain may have been important to the evolution of the Coccidioides genomes, it is confounded by gene loss along the U. reesii lineage. Utilizing H. capsulatum as an outgroup, a parsimony-based analysis of 4460 orthologs reveals that the U. reesii lineage experienced a greater rate of gene turnover (1075 gains and 399 losses) than the Coccidioides lineages did (280–291 gains and 109 losses). However, to account for annotation differences, Coccidioides gene gains and losses were only considered if both species had at least one copy of the ortholog. Thus, these findings may represent an underestimate of the total gene turnover rate in either species. Also, given the phylogenetic distances involved and the use of only four taxa, gene gains may alternatively result from independent, parallel loss of ancestral genes in both the sister and outgroup lineages.
The endosporulating spherule stage of the Coccidioides lifecycle is a unique Ascomycete phenotype and is necessary for pathogenicity (Rappleye et al. 2006). Of the 1201 genes identified as having spherule-specific expression (Methods), 93 are genes that have been gained in the Coccidioides lineage since it diverged with U. reesii (Supplemental Table 10). These gained spherule-specific genes are enriched for several functions, including generation of precursor metabolites and energy (P = 0.002), mitochondrion-related (P = 0.048), and cation-transporting ATPase activity (P = 0.025). In addition, 24 of these 93 genes exhibit signal peptides or membrane anchors, suggesting that they may be involved in cell surface-/membrane-related biology.
Positive natural selection of Onygenales proteins
Coccidioides proteins subject to positive natural selection may have contributed to the organism's adaptation to life within an immunocompetent mammalian host. The nonsynonymous (dN) to synonymous (dS) ratio is conventionally calculated to identify proteins with signatures of positive selection (Nielsen et al. 2005); however, the median dS between Coccidioides and its closest genome sequenced relative, U. reesii, is 1.60 (calculated from 5953 three-way orthologs), indicating that synonymous nucleotide mutations are saturated and the taxa are too diverged to use this methodology with confidence (Supplemental Fig. 11). Alternatively, a relative rates test can be used to detect lineage-specific accelerations in the amino acid substitution rate, which can result from a change in selection pressure. Using H. capsulatum as an outgroup and maximum likelihood analysis to infer ancestral character states, a relative rates test of 4460 four-way conserved orthologs identifies 67 Coccidioides genes that have evolved with a significantly faster amino acid substitution rate since divergence with U. reesii (P < 0.05; Supplemental Table 12). These genes are functionally enriched for biopolymer metabolic processes (P = 0.017) and RNA metabolic process (P = 0.022). One noteworthy gene (CIMG_07738) is homologous to the expression library immunization antigen 1 protein, which, when used as a vaccine, has been shown to provide protection against coccidioidomycosis to BALB/c mice (Ivey et al. 2003).
Identifying genes subject to positive natural selection since two closely related pathogens diverged may identify antigenic compounds (Esteves et al. 2008). The low median dS value of 0.023 calculated from the 6763 orthologs conserved between C. posadasii and C. immitis indicates that the taxa diverged recently enough to identify the signature of positive selection via comparisons of dN and dS. As observed in other organisms (Nielsen et al. 2005), the median dN/dS value is low (0.25), suggesting that most genes have evolved under purifying selection (Supplemental Fig. 13). However, there are 57 genes that exhibit signatures of positive natural selection with dN/dS significantly greater than 1 (P < 0.05; Supplemental Table 14). These genes are enriched for several GO categories, including metabolic process (P = 2.6 × 10−4), phosphorylation (P = 0.012), and S-adenosylmethionine-dependent methyltransferase activity (P = 9.7 × 10−3). In addition, while in vivo analysis must confirm host immune system interaction, five secreted proteins of unknown function exhibit signatures of positive selection (CIMG_03148, CIMG_06700, CIMG_02887, CIMG_09110, CIMG_05660), and thus may be good candidates for functional analysis and vaccine discovery.
Proline-rich antigens
Prior work has shown that a recombinant divalent vaccine of the Coccidioides proline-rich protein paralogs Prp1 (also known as antigen 2 or the proline-rich antigen, rAG2/Pra) and Prp2 provides better protection than either protein alone (Herr et al. 2007). It is believed that this improved performance is due to host exposure to a more heterogeneous set of T-cell epitopes, which enhances the Th1 response to infection and provides a more robust and sustained immune response. Searching the C. posadasii genome for additional Prp homologs identifies six additional genes in this family, which were subjected to the ProPred algorithm to predict the presence of MHC class II epitopes. Of these six additional Prp paralogs, Prp5, −7, and −8 contain the highest number of promiscuous T-cell epitopes, which bind to numerous histocompatibility alleles (Table 3; Supplemental Fig. 15). In addition, Prp5 has the highest epitope ratio (epitopes/kilodalton) and shows the greatest reactivity in IFNG ELISPOT assays (Supplemental Fig. 16). As the best-predicted vaccine candidate, it is proposed for inclusion in a trivalent vaccine (Prp1 + Prp2 + Prp5) against coccidioidomycosis.
Human MHC class II binding epitope prediction of C. posadasii PRP proteins
Discussion
This study reports on the genome sequencing and comparison of the primary human pathogens C. immitis and C. posadasii; a recently diverged nonpathogenic relative, U. reesii; and a more diverged pathogen from the Onygenales, H. capsulatum (Supplemental Fig. 17). These sequences reveal that despite having a size similar to other filamentous Ascomycetes, Onygenales genomes are unusual in their chromosome number. For example, whereas most filamentous Ascomycetes have between seven and eight chromosomes, the Coccidioides genomes only have five. However, the chromosome structure of Coccidioides is similar to other Ascomycetes (Cuomo et al. 2007; Fedorova et al. 2008) in that they contain species-specific “genomic islands,” many of which are located in chromosomal subtelomeric regions. Interestingly, eight of these islands occupy identical syntenic positions in C. posadasii and C. immitis, suggesting that they originate through nonrandom processes.
Repetitive genomic sequences in Coccidioides exhibit a biased nucleotide and dinucleotide composition, a phenomenon observed in other filamentous fungi that are subject to RIP (repeat induced point-mutation). RIP is a mutational process that detects duplicated DNA and introduces C:G to T:A transitions into the repeats (Selker and Garrett 1988; Montiel et al. 2006). Experimental observations in several Sordariomycete fungi (Selker and Garrett 1988; Ikeda et al. 2002; Cuomo et al. 2007) suggest that sequences subject to RIP exhibit a paucity of CpA dinucleotides, which are preferential targets of the mutational process (Galagan and Selker 2004). Similar to the Afut1 transposon of the Eurotiomycete A. fumigatus (Neuveglise et al. 1996), prior analysis of C. posadasii crypton transposons identified a RIP-like signature in that these repetitive sequences exhibit a paucity of CpG dinucleotides (Gardiner and Howlett 2006). The current study's survey of the Coccidioides genomes for biased dinucleotide frequencies extends this finding by identifying a genome-wide correlation between a paucity of CpG dinucleotides and repetitive content (Fig. 1). While such a correlation is suggestive of RIP, because the canonical RIP signature of a skewed CpA distribution is not observed, it cannot be strongly concluded that RIP, at least as it is defined for Sordariomycetes, has shaped the evolution of Coccidioides repeats. The repeat-associated CpG bias observed in Coccidioides could be the result of a wholly different mutational process, or perhaps RIP has evolved a heightened preference for CpG dinucleotides as a mutational target in these fungi.
Comparing these newly sequenced genomes across the Pezizomycotina enabled detection of substantial gene family size changes in Coccidioides and related fungi. In Coccidioides, families of plant cell wall degrading enzymes are reduced in number compared with those of fungi that are known to decay plant matter. In contrast to the Aspergillus species, for example, Coccidioides exhibits few or no cellulases, cutinases, tannases, or pectinesterases and exhibits reductions in numbers of genes involved in sugar metabolism (Supplemental Table 7). Coupled with these losses in plant degrading enzymes are expansions of protease families in Coccidioides relative to other filamentous Ascomycetes, the most notable being keratinase. In sum, these changes suggest that Coccidioides species are not typical soil fungi in that they maintain a close association with keratin-rich animals both during infection of a living host and after the host has died by growing through the carcass as mycelium. Additional evidence about lifecycle may be found in the deficit of dehydrogenases and HET proteins in Coccidioides relative to Aspergillus, which suggests that Coccidioides utilizes fewer metabolites and encounters fewer genetically distinct individuals than their soil saprobe counterparts. Such speculation is consistent with previous research showing an association of Coccidioides with living animals (Emmons 1942), and the difficulty of recovering Coccidioides species from soil, due to its extremely patchy distribution (Maddy 1967; Greene et al. 2000).
The gene family size changes observed in Coccidioides are conserved in U. reesii. The most parsimonious explanation of this observation is that adaptation by gene family change related to the shift from plant to animal substrate must have occurred before the divergence of these two taxa. In contrast, another pathogenic member of the Onygenales, H. capsulatum, does not share the protease expansions observed in Coccidioides, though it has experienced a similar reduction of plant decay enzymes. Although Coccidioides and Histoplasma are both environmentally acquired dimorphic pathogens of mammals, the niche of their conidia-producing lifecycle stage is different. Unlike Coccidioides, whose free-living stage appears to be limited to association with a host carcass, H. capsulatum grows in the environment in association with bird and bat guano. As a result, Histoplasma may be equipped to survive in host excrement and, unlike Coccidioides, may be able to propagate infection without requiring host death. If correct, this thinking suggests that the restricted association to dead hosts of Coccidioides evolved after the divergence of H. capsulatum from the Coccidioides/Uncinocarpus lineage.
The similarities in gene family size between the Coccidioides species and U. reesii suggest that gene family expansion and contraction alone are not responsible for the pathogenic phenotype observed in Coccidioides. Although U. reesii can grow on animal tissue and can be cultured from animal organs and hair (Sigler and Carmichael 1976), this organism has never been known to cause disease, cannot be grown at 37°C, and is only known to grow in a filamentous morphology. Therefore, genomic differences other than gene family size must account for the phenotypic differences observed between Coccidioides and U. reesii, including pathogenesis. An analysis of gene gain and loss and of genes evolving under positive selection identified several proteins with functions that suggest involvement in pathogenesis and thus should be considered candidates for in vivo functional analysis.
First, metabolism-related proteins may contribute to the infectious life history of Coccidioides. For example, following divergence from U. reesii, Coccidioides species either acquired or retained a suite of genes enriched for heme/tetrapyrrole binding (P = 3.5 × 10−4), an attribute of a class of proteins shown to be important for scavenging iron within a host (Howard 1999). In addition, the set of Coccidioides-specific genes with spherule-specific expression are enriched for the generation of precursor metabolites and energy (P = 0.002). Among this set is a key enzyme involved in allantoin metabolism, a nitrogen-rich compound produced as a by-product of free-radical attack in mammals that is utilized by some pathogens when nitrogen is limited (Divon and Fluhr 2007). These findings suggest that Coccidioides has acquired genes that enable the fungus to grow on metabolites found within a host, such as iron and nitrogen.
Second, membrane-related genes have been important in the evolution of Coccidioides and may contribute to its infectious phenotype. For example, the genes uniquely gained in Coccidioides are enriched for integral membrane function (P = 1.7 × 10−4). Of those that are also uniquely expressed in the parasitic morphology, roughly one-third exhibit signal peptides or signal anchors, indicating they may be involved in cell surface or membrane biology. Additionally, among the proteins found to be rapidly evolving in the Coccidioides lineage are a transmembrane amino acid transporter and a major facilitator superfamily transporter, homologs of which provide Candida albicans access to the limiting metabolite nitrogen during host infection (Biswas and Monideepa 2003) and provide multidrug resistance to azoles and other fungicides (Del Sorbo et al. 2000), respectively. Also included in this group of genes is a lipase, which is a member of a protein family implicated in several fungal virulence mechanisms involving the cell membrane (Ghannoum 2000). This finding suggests that lipases may be important to the adaptation of Coccidioides, possibly by contributing to the development of the membrane-rich endosporulating spherules, the morphology required for disseminated infection. Because mutations that prevent endospore formation result in avirulence, proteins involved in cell wall and membrane differentiation during endosporulation may warrant further consideration as potential drug targets.
Finally, secondary metabolism may have contributed to the evolution of virulence in Coccidioides. A gene that shares homology with the mycotoxin Asp-hemolysin was either gained or retained in Coccidioides relative to other Onygenales, and EST data suggest it is uniquely expressed in the pathogenic morphology. In addition, rapidly evolving Coccidioides proteins are enriched for S-adenosylmethionine-dependent methyltranserase activity. In Aspergillus flavus and Aspergillus parasiticus, this function is responsible for the penultimate step in aflatoxin biosynthesis. While experimental verification is required, these findings suggest that Coccidioides may produce secondary metabolites, such as mycotoxins, that facilitate the infection process.
Taken together, these findings suggest a timeline for the evolution of the pathogenic phenotype of Coccidioides (Fig. 2). The first step in this evolutionary transition was the shift from a nutritional association with plants to animals. This event occurred before Coccidioides and U. reesii diverged and is revealed through analysis of gene family size. Because U. reesii and other closely related fungi grow in association with animals but are not known to cause disease, it is most parsimonious to assume that virulence was recently and uniquely acquired by Coccidioides and, thus, that this shift in nutritional association did not directly yield this phenotype. Rather, the common ancestor of Coccidioides and U. reesii likely grew on dead or decaying animal matter or on external surfaces of an animal host, such as hair. However, once this association with animals was established, ongoing lineage-specific evolution in Coccidioides—most notably in metabolism, membrane, and secondary metabolism genes—enabled the adaptation to survive and grow within an animal host, ultimately yielding disease. The two Coccidioides species diverged ∼5.1 Mya, well before humans arrived in the New World (Pitulko et al. 2004), so this evolutionary transition to a pathogenic phenotype likely involved primarily rodent hosts and not humans.
In summary, the results of this study suggest that the pathogenic phenotype of Coccidioides is the result of adaptive changes to existing genes and the acquisition of small numbers of new genes that began with a shift from plant-based to animal-based nutrition. Though the methodology presented here identifies significant evolutionary changes in the Coccidioides lineage, improving the phylogenetic resolution during relatively recent Coccidioides evolution and the practical knowledge regarding its functional genetics and ecology would facilitate a superior understanding of the organisms' life history and evolution. Future studies that engage in population-level analyses within Coccidioides, that compare Coccidioides to the genomes of other closely related, nonpathogenic Onygenales, and that clarify the functional genetics and ecology of Coccidioides will be necessary to test the proposed hypotheses and, thus, improve the understanding of the evolutionary origins and treatment of coccidioidomycosis.
Methods
Genome sequences and annotations utilized
The genome for C. posadasii strain C735ΔSOWgp (March 2006 release, http://www.tigr.org/tdb/e2k1/cpa1/) was sequenced at 8× coverage via Sanger shotgun sequencing by the J. Craig Venter Institute (formerly The Institute for Genome Research). The AAT alignment package identified genome sequence homologs of proteins in the NCBI nonredundant database and custom fungal protein databases. In addition, tigrscans and glimmerHMM were trained via high-confidence, manually curated gene models to predict genes ab initio. Also, PASA assembled and aligned 53,664 ESTs, of which 26,201 were derived from mycelium and 27,463 of which were derived from spherules (Supplemental Fig. 18; Haas et al. 2003). Combiner used this evidence to produce a final set of annotation predictions (Allen et al. 2004), which were then functionally annotated via comparison to the following protein, domain, and profile databases: NR, Pfam, TIGRfam HMMs, Prosite, and Interpro. In addition, SignalP, TMHMM, and TargetP were used to predict protein membrane localization.
The C. immitis strain RS (June 2008, AAEC02000000), U. reesii strain 1704 (May 2006 release, AAIW01000000), and H. capsulatum strain WU24 (May 2006; AAJI01000000) genome sequences were generated at 14.4×, 5×, and 7×, respectively, by the Broad Institute using Sanger shotgun sequencing as described by Galagan et al. (2003). The U. reesii and H. capsulatum annotations were generated using automated pipelines. The C. immitis annotation employed 65,754 C. immitis RS ESTs, including from 22,382 mycelia and 43,372 from spherules, and was manually reviewed.
AUGUSTUS was used to predict genes ab initio in the following genome sequences: A. flavus strain NRRL3357 (AAIH00000000), Blastomyces dermatitidis strain ATCC 26199 (http://genome.wustl.edu/pub/organism/Fungi/Blastomyces_dermatitidis/assembly/Blastomyces_dermatitidis-3.0/), H. capsulatum strain 186AR (http://genome.wustl.edu/pub/organism/Fungi/Histoplasma_capsulatum/assembly/Histoplasma_capsulatum_G186AR-3.0/), H. capsulatum strain 217B (http://genome.wustl.edu/pub/organism/Fungi/Histoplasma_capsulatum/assembly/Histoplasma_capsulatum_G217B-3.0/), Paracoccidioides brasiliensis strain pb03 (http://www.broad.mit.edu/annotation/genome/paracoccidioides_brasiliensis/MultiHome.html), and Penicillium marneffei strain ATCC 18224 (ABAR00000000). Genome sequence and annotation data for A. clavatus strain NRRL (AAKD00000000), A. fumigatus strain Af293 (Nierman et al. 2005), Aspergillus nidulans strain A4 (Galagan et al. 2005), Aspergillus niger strain CBS51388 (Pel et al. 2007), Aspergillus oryzae strain RIB40 (Machida et al. 2005), Aspergillus terreus strain NIH2642 (AABT01000000), Fusarium graminearum strain PH-1 (Cuomo et al. 2007), Neurospora crassa strain OR74A (Galagan et al. 2003), and Sclerotinia sclerotiorum strain 1980 (AAGT01000000) were downloaded from GenBank.
Ortholog detection and alignment
Several orthology data sets were generated including 18-way clusters among the Pezizomycotina, four-way orthology clusters between Onygenalean fungi C. immitis, C. posadasii, U. reesii, and H. capsulatum; three-way clusters between C. immitis, C. posadasii, and U. reesii; and pairwise orthologs between the two Coccidioides species. For each data set, all predicted protein sequences from the appropriate genomes were searched against each other with BLASTP and clustered into orthologous groups using OrthoMCL (Li et al. 2003) with the following criteria: the alignment must cover 60% of the query sequence and the E-value must be less than 10−5. Single-copy orthologs were identified as the clusters with exactly one member per species. Multiple sequence alignments were constructed with MUSCLE (Edgar 2004).
Phylogenetic analysis
Phylogenetic relationships were determined for the following 18 taxa: C. immitis, C. posadasii, U. reesii, H. capsulatum (strains 186R, 217B, and WU24), B. dermatitidis, P. brasiliensis, A. fumigatus, A. clavatus, A. flavus, A. niger, A. oryzae, A. terreus, A. nidulans, P. marneffei, S. sclerotiorum, and F. graminearum. Single-copy orthologs from OrthoMCL clusters were aligned with ProbCons (Do et al. 2005). Alignments were pruned with Gblocks (Castresana 2000), and data were combined into a single alignment file containing 823,457 characters. Eighteen random subselections were chosen without replacement and used to construct phylogenetic trees with three methods: neighbor-joining (NJTREE), Bayesian with MrBayes (Ronquist and Huelsenbeck 2003), and maximum likelihood with RAxML (Stamatakis et al. 2005). Divergence time estimation was performed using nonparametric rate smoothing (NPRS) (Sanderson 2003) with the maximum likelihood tree as a starting point and calibration of 215 Myr for the Pezizomycotina origin.
Repetitive content and chromosome structure
The C. immitis and C. posadasii genomes were divided into 1-kb nonoverlapping windows and then aligned to one another via BLASTN. Window copy number was calculated by counting the number of detected homologs greater than 500 bp in length for each window and were subsequently divided into three copy number frequency classes: small (n < 6), medium (n = 6–20), and large (n > 20). Repetitive sequence was partially characterized by searching each window against the Repbase Fungal repeat database using TBLASTN and an E-value cutoff of 1 × 10−2. Windows with no detectable homologs were considered species specific.
Lineage-specific gene gain and loss
A parsimony analysis of an ortholog's phylogenetic profile identified Coccidioides and U. reesii lineage-specific gene gain and loss. H. capsulatum was used to infer the ortholog's ancestral state. A gene was considered lost along a lineage if the taxon in question was missing an ortholog shared between the sister and outgroup taxa. Conversely, a gene was considered gained along a lineage if no other taxon shared the ortholog in question. For the Coccidioides taxa, a gene was only considered if the ortholog's phylogenetic profile was the same in both species.
The 53,664 C. posadasii ESTs (26,201 from mycelium and 27,463 derived from in vitro spherules) and the 65,754 C. immitis ESTs (22,382 from mycelia and 43,372 from in vitro spherules) were aligned to annotated genes via BLAT (Kent 2002). Evaluating the morphology from which the aligned ESTs were derived enabled classification of gene expression condition. Putative spherule-specific genes were thus identified as those that matched at least one spherule derived EST and no mycelial ESTs.
Gene family expansion and contraction analysis
Gene family size was determined for the following genome sequenced Pezizomycetes: A. terreus, A. oryzea, A. niger, A. fumigatus, A. nidulans, C. posadasii, C. immitis, U. reesii, H. capsulatum, Stagonospora nodorum, Chaetomium globosum, Podospora anserina, N. crassa, Magnaporthe grisea, F. graminearum, F. verticillioides, and F. solani. For each genome sequence, Pfam protein domains (Finn et al. 2008) were identified via HMMER using a cutoff value of E < 10−2, and the number of genes containing at least one copy of each Pfam protein domain was determined. Using a multigene phylogeny, gene family sizes were associated with each taxon to determine the magnitude of gene family expansions and contractions. CAFE was used to detect significant gene family size changes between any two lineages as defined in De Bie et al. (2006). Gene family counts for internal branches were calculated by averaging the counts for the daughter branches of the lineage in question.
Detection of positive selection
The M0 model in PAML (v.3.15) (Yang 1997) was used to identify protein substitution rates of orthologs in clusters containing single representatives from C. immitis, C. posadasii, U. reesii, and the outgroup H. capsulatum. These rates were mapped onto the species phylogeny and a χ2 approximation was used to determine if any two lineages displayed significantly different substitution rates (P < 0.05), after accounting for multiple testing via a Bonferroni correction.
PAML was also used to estimate ω, the ratio of the dN and dS substitution rates (ω = dN/dS), for all orthologous coding sequence pairs shared between C. immitis and C. posadasii. Values of ω = 1 indicate neutral evolution, while values of ω significantly less and greater than 1 indicate purifying and positive directional selection, respectively. Two evolutionary models were used to estimate ω—a null model where ω was fixed (ω = 1) and another where ω was unconstrained and estimated from the data—and a likelihood ratio test of each model's maximum likelihood score identified orthologs for which the null hypothesis could be rejected (P < 0.05) after correcting for multiple tests using Q-value (P < 0.05) (Storey and Tibshirani 2003). Processing of alignments, trees, and PAML results was handled through Perl scripts utilizing the modules in the BioPerl package (Stajich et al. 2002).
GO enrichment analysis
Pfam protein domains were used to assign proteins to GO categories via PFAM2GO data provided by the GO consortium (Ashburner et al. 2000). Proteins with multiple domains corresponding to different functions were allowed to belong to multiple GO categories. A particular GO function was considered enriched if its representation among a selected subset of a taxon's genes is significantly greater than its representation across the taxon's genome (P < 0.05 after a Bonferroni multiple test correction).
Acknowledgments
Genome sequencing at the Broad Institute was supported by both NHGRI and NIAID through the Broad Institute's Fungal Genome Initiative and its Microbial Sequencing Center. Sequencing of the C. posadasii C735 genome at TIGR and JCVI was supported by NIAID VO1 AI50910 (M.J.G. and G.T.C.). J.E.S. was supported by a postdoctoral fellowship from the Miller Institute for Basic Research at U.C., Berkeley. T.J.S. was supported by the Chang-lin Tien graduate fellowship at U.C., Berkeley. All phases of analysis were supported by NIH/NIAID R01AI70891 (J.W.T. and G.T.C.).
References
- ↵JE Allen M Pertea SL Salzberg . 2004. Computational gene prediction using multiple sources of evidence. Genome Res 14: 142– 148
- ↵M Ashburner CA Ball JA Blake D Botstein H Butler JM Cherry AP Davis K Dolinski SS Dwight JT Eppig 2000. Gene Ontology: Tool for the unification of biology. Nat Genet 25: 25– 29
- ↵BM Barker KA Jewell S Kroken MJ Orbach . 2007. The population biology of Coccidioides: Epidemiologic implications for disease outbreaks. Ann N Y Acad Sci 1111: 147– 163
- ↵S Biswas R Monideepa . 2003. N-acetylglucosamine–inducible CaGAP1 encodes a general amino acid permease which coordinates external nitrogen source response and morphogenesis in Candida albicans. Microbiology 149: 2597– 2608
- ↵L Cao H Tan Y Liu X Xue S Zhou . 2008. Characterization of a new keratinolytic Trichoderma atroviride strain F6 that completely degrades native chicken feather. Lett Appl Microbiol 46: 389– 394
- ↵J Castresana . 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540– 552
- ↵S Censini C Lange ZY Xiang JE Crabtree P Ghiara M Borodovsky R Rappuoli A Covacci . 1996. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci 93: 14648– 14653
- ↵CA Cuomo U Gueldener JR Xu F Trail BG Turgeon A Di Pietro JD Walton LJ Ma SE Baker M Rep 2007. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317: 1400– 1402
- ↵BA da Silva ALS dos Santos E Barreto-Bergter MR Pinto . 2006. Extracellular peptidase in the fungal pathogen Pseudallescheria boydii. Curr Microbiol 53: 18– 22
- ↵T De Bie N Cristianini JP Demuth MW Hahn . 2006. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 22: 1269– 1271
- ↵G Del Sorbo HJ Schoonbeek MA De Waard . 2000. Fungal transporters involved in efflux of natural toxic compounds and fungicides. Fungal Genet Biol 30: 1– 15
- ↵F Descamps . 2005. Isolation and characterization of a Microsporum canis gene family encoding three subtilisin-like serine proteases and contribution to the study of their role in the host-fungus relationship. Ann Med Vet 149: 11– 15
- ↵HH Divon R Fluhr . 2007. Nutrition acquisition strategies during fungal infection of plants. FEMS Microbiol Lett 266: 65– 74
- ↵DM Dixon . 2001. Coccidioides immitis as a select agent of bioterrorism. J Appl Microbiol 91: 602– 605
- ↵CB Do MSP Mahabhashyam M Brudno S Batzoglou . 2005. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15: 330– 340
- ↵RC Edgar . 2004. MUSCLE: Multiple sequence alignment with high accuracy and high-throughput. Nucleic Acids Res 32: 1792– 1797
- ↵CW Emmons . 1942. Isolation of Coccidioides from soil and rodents. Public Health Rep 57: 109– 111
- ↵E Espagne P Balhadere ML Penin C Barreau B Turcq . 2002. HET-E and HET-D belong to a new subfamily of WD40 proteins involved in vegetative incompatibility specificity in the fungus Podospora anserina. Genetics 161: 71– 81
- ↵PJ Esteves J Abrantes M Carneiro A Muller G Thompson W van der Loo . 2008. Detection of positive selection in the major capsid protein VP60 of the rabbit haemorrhagic disease virus (RHDV). Virus Res 137: 253– 256
- ↵ND Fedorova N Khaldi VS Joardar R Maiti P Amedeo MJ Anderson J Crabtree JC Silva JH Badger A Albarraq 2008. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4: e1000046– 10.1371/journal.pgen.1000046
- ↵RD Finn J Tate J Mistry PC Coggill SJ Sammut HR Hotz G Ceric K Forslund SR Eddy ELL Sonnhammer 2008. The Pfam protein families database. Nucleic Acids Res 36: D281– D288
- ↵MC Fisher G Koenig TJ White JW Taylor . 2000. A test for concordance between the multilocus genealogies of genes and microsatellites in the pathogenic fungus Coccidioides immitis. Mol Biol Evol 17: 1164– 1174
- ↵FS Fisher MW Bultman SM Johnson D Pappagianis E Zaborsky . 2007. Coccidioides niches and habitat parameters in the Southwestern United States—a matter of scale. Ann N Y Acad Sci 1111: 47– 72
- ↵JE Galagan EU Selker . 2004. RIP: The evolutionary cost of genome defense. Trends Genet 20: 417– 423
- ↵JE Galagan SE Calvo KA Borkovich EU Selker ND Read D Jaffe W FitzHugh LJ Ma S Smirnov S Purcell 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422: 859– 868
- ↵JE Galagan SE Calvo C Cuomo LJ Ma JR Wortman S Batzoglou SI Lee M Basturkmen CC Spevak J Clutterbuck 2005. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438: 1105– 1115
- ↵J Galgiani NM Ampel J Blair A Catanzaro . 2005. Coccidioidomycosis. Clin Infect Dis 4: 1217– 1223
- ↵D Gardiner B Howlett . 2006. Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol Lett 248: 241– 248
- ↵DM Geiser C Gueidan J Miadlikowska F Lutzoni F Kauff V Hofstetter E Fraker CL Schoch L Tibell WA Untereiner 2006. Eurotiomycetes: Eurotiomycetidae and Chaetothyriomycetidae. Mycologia 98: 1053– 1064
- ↵MA Ghannoum . 2000. Potential role of phospholipases in virulence and fungal pathogenesis. Clin Microbiol Rev 13: 122– 143
- ↵DR Greene G Koenig MC Fisher JW Taylor . 2000. Soil isolation and molecular identification of Coccidioides immitis. Mycologia 92: 406– 410
- ↵BJ Haas AL Delcher SM Mount JR Wortman RK Smith LI Hannick R Maiti CM Ronning DB Rusch CD Town 2003. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31: 5654– 5666
- ↵RF Hector R Laniado-Laborin . 2005. Coccidioidomycosis—a fungal disease of the Americas. PLoS Med 2: e2– 10.1371/journal.pmed.0020002
- ↵R Hector GW Rutherford . 2007. The public health need and present status of a vaccine for the prevention of Coccidioidomycosis. Ann N Y Acad Sci 1111: 259– 268
- ↵IR Henderson JP Nataro . 2001. Virulence functions of autotransporter proteins. Infect Immun 69: 1231– 1243
- ↵JL Hernandez S Echevarria A GarciaValtuille F Mazorra R Salesa . 1997. Atypical coccidioidomycosis in an AIDS patient successfully treated with fluconazole. Eur J Clin Microbiol Infect Dis 16: 592– 594
- ↵RA Herr CY Hung GT Cole . 2007. Evaluation of two homologous proline-rich proteins of Coccidioides posadasii as candidate vaccines against coccidioidomycosis. Infect Immun 75: 5777– 5787
- ↵DH Howard . 1999. Acquisition, transport, and storage of iron by pathogenic fungi. Clin Microbiol Rev 12: 394– 404
- ↵CY Hung JJ Yu KR Seshan U Reichard GT Cole . 2002. A parasitic phase-specific adhesin of Coccidioides immitis contributes to the virulence of this respiratory fungal pathogen. Infect Immun 70: 3443– 3456
- ↵CY Hung KR Seshan JJ Yu R Schaller JM Xue V Basrur MJ Gardner GT Cole . 2005. A metalloproteinase of Coccidioides posadasii contributes to evasion of host detection. Infect Immun 73: 6689– 6703
- ↵K-I Ikeda H Nakayashiki T Kataoka H Tamba Y Hashimoto Y Tosa S Mayama . 2002. Repeat-induced point mutation (RIP) in Magnaporthe grisea: Implications for its sexual cycle in the natural field context. Mol Microbiol 45: 1355– 1364
- ↵FD Ivey DM Magee MD Woitaske SA Johnston RA Cox . 2003. Identification of a protective antigen of Coccidioides immitis by expression library immunization. Vaccine 21: 4359– 4367
- ↵TY James F Kauff CL Schoch PB Matheny V Hofstetter CJ Cox G Celio C Gueidan E Fraker J Miadlikowska 2006. Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature 443: 818– 822
- ↵WJ Kent . 2002. BLAT: The BLAST-Like Alignment Tool. Genome Res 12: 656– 664
- ↵V Koufopanou A Burt JW Taylor . 1998. Concordance of gene genealogies reveals reproductive isolation in the pathogenic fungus Coccidioides immitis. Proc Natl Acad Sci 95: 5478– 5482
- ↵JG Lawrence . 2005. Common themes in the genome strategies of pathogens. Curr Opin Genet Dev 15: 584– 588
- ↵L Li CJ Stoeckert DS Roos . 2003. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178– 2189
- ↵M Machida K Asai M Sano T Tanaka T Kumagai G Terai KI Kusumoto T Arima O Akita Y Kashiwagi 2005. Genome sequencing and analysis of Aspergillus oryzae. Nature 438: 1157– 1161
- ↵KT Maddy . 1967. Epidemiology and ecology of deep mycoses of man and animals. Arch Dermatol 96: 409– 417
- ↵M Monod S Capoccia B Lechenne C Zaugg M Holdom O Jousson . 2002. Secreted proteases from pathogenic fungi. Int J Med Microbiol 292: 405– 419
- ↵MD Montiel HA Lee DB Archer . 2006. Evidence of RIP (repeat-induced point mutation) in transposase sequences of Aspergillus oryzae. Fungal Genet Biol 43: 439– 445
- ↵C Neuveglise J Sarfati JP Latge S Paris . 1996. Afut1, a retrotransposon-like element from Aspergillus fumigatus. Nucleic Acids Res 24: 1428– 1434
- ↵R Nielsen C Bustamante AG Clark S Glanowski TB Sackton MJ Hubisz A Fledel-Alon DM Tanenbaum D Civello TJ White 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3: 976– 985
- ↵WC Nierman A Pain MJ Anderson JR Wortman HS Kim J Arroyo M Berriman K Abe DB Archer C Bermejo 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438: 1151– 1156
- ↵S Pan GT Cole . 1992. Electrophoretic karyotypes of clinical isolates of Coccidioides immitis. Infect Immun 60: 4872– 4880
- ↵D Papagiannis . 1967. Epidemiological aspects of respiratory mycotic infections. Bacteriol Rev 31: 25– 34
- ↵HJ Pel JH de Winde DB Archer PS Dyer G Hofmann PJ Schaap G Turner RP de Vries R Albang K Albermann 2007. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25: 221– 231
- ↵VV Pitulko PA Nikolsky EY Girya AE Basilyan VE Tumskoy SA Koulakov SN Astakhov EY Pavlova MA Anisimov . 2004. The Yana RHS site: Humans in the arctic before the last glacial maximum. Science 303: 52– 56
- ↵CA Rappleye WE Goldman . 2006. Defining virulence genes in the dimorphic fungi. Annu Rev Microbiol 60: 281– 303
- ↵F Ronquist JP Huelsenbeck . 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572– 1574
- ↵MJ Sanderson . 2003. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19: 301– 302
- ↵R Segers TM Butt JH Carder JN Keen BR Kerry JF Peberdy . 1999. The subtilisins of fungal pathogens of insects, nematodes and plants: Distribution and variation. Mycol Res 103: 395– 402
- ↵EU Selker PW Garrett . 1988. DNA-sequence duplications trigger gene inactivation in Neurospora crassa. Proc Natl Acad Sci 85: 6870– 6874
- ↵L Sigler JK Carmichael . 1976. Taxonomy of malbranchea and some other hyphomycetes with Arthroconidia. Mycotaxon 4: 349– 488
- ↵JE Stajich D Block K Boulez SE Brenner SA Chervitz C Dagdigian G Fuellen JGR Gilbert I Korf H Lapp 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res 12: 1611– 1618
- ↵A Stamatakis T Ludwig H Meier . 2005. RAxML-III: A fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21: 456– 463
- ↵JD Storey R Tibshirani . 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440– 9445
- ↵JW Taylor ML Berbee . 2006. Dating divergences in the fungal tree of life: Review and new analyses. Mycologia 98: 838– 849
- ↵WA Untereiner JA Scott FA Naveau L Sigler J Bachewich A Angus . 2004. The Ajellomycetaceae, a new family of vertebrate-associated Onygenales. Mycologia 96: 812– 821
- ↵ZH Yang . 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13: 555– 556
Notes
[3] [Supplemental material is available online at http://www.genome.org. The sequence data for C. immitis, C. posadasii, H. capsulatum, and U. reesii have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) under accession nos. AAEC02000000, ACFW00000000, AAJI01000000, and AAIW01000000, respectively.]
[4] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.087551.108.