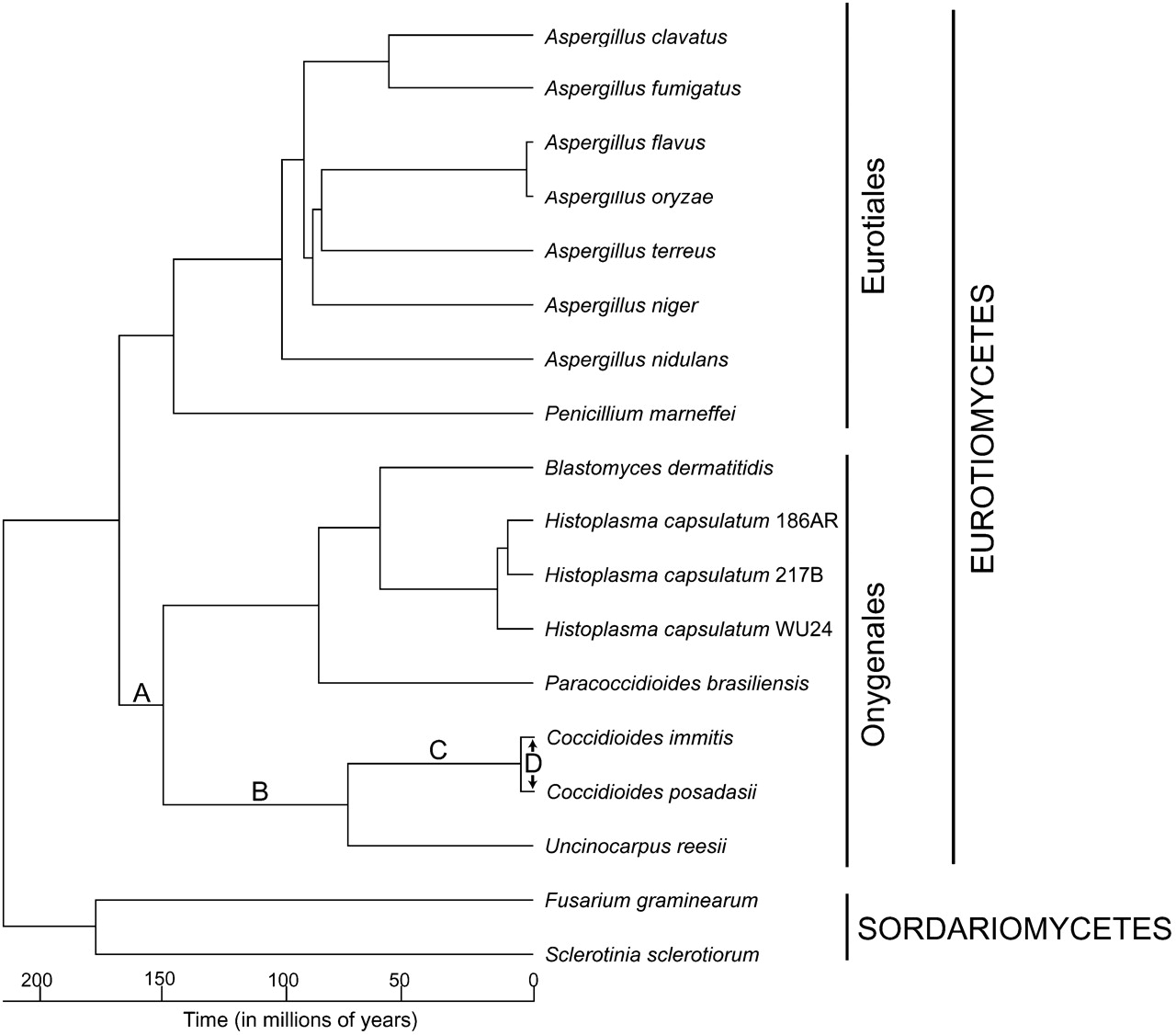
A timeline of genomic changes that underlie the evolution of pathogenicity in Coccidioides. A RAxML-generated maximum likelihood phylogeny based on an alignment of 1148 concatenated genes that comprise 46,890 unambiguously aligned amino acid sites following the removal of gaps was used as a starting point to perform nonparametric rate smoothing. Divergence times were subsequently estimated from the smoothed phylogeny by applying a calibration of 215 Mya for the Pezizomycotina origin as in the method of Taylor and Berbee (2006). The significant evolutionary events identified in the hierarchical comparative genomic analysis of the Coccidioides lineage are overlaid onto the phylogeny. Taken together, they suggest that a gradual progression of evolutionary events ultimately yielded the pathogenic phenotype of Coccidioides. (A) Gene family reductions associated with the metabolism of plant material were coupled with a growth substrate transition from plant to animal material. (B) Subsequent expansions in proteases and keratinases led to a nutritional association with mammals in a nonpathogenic fashion. (C) The acquisition and adaptation of genes involved in metabolism, membrane biology, and mycotoxin production led to metabolic and morphological phenotypes that enabled survival within a live host, ultimately resulting in disease. (D) Secreted proteins, metabolism genes, and secondary metabolism genes that are shared between the two Coccidioides species have been subject to positive selection since they diverged. This suggests that Coccidioides may experience on-going adaptation in response to host immune system selection pressures.