
Efficient mapping of accurate long reads in minimizer space with mapquik
- 1Computer Science and Artificial Intelligence Laboratory (CSAIL), Massachusetts Institute of Technology (MIT), Cambridge, Massachusetts 02139, USA;
- 2Department of Mathematics, Massachusetts Institute of Technology (MIT), Cambridge, Massachusetts 02139, USA;
- 3Department of Mathematics, Science for Life Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden;
- 4Department of Computer Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 5Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 6Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 7Department of Computational Biology, Institut Pasteur, 75015 Paris, France
Abstract
DNA sequencing data continue to progress toward longer reads with increasingly lower sequencing error rates. We focus on the
critical problem of mapping, or aligning, low-divergence sequences from long reads (e.g., Pacific Biosciences [PacBio] HiFi)
to a reference genome, which poses challenges in terms of accuracy and computational resources when using cutting-edge read
mapping approaches that are designed for all types of alignments. A natural idea would be to optimize efficiency with longer
seeds to reduce the probability of extraneous matches; however, contiguous exact seeds quickly reach a sensitivity limit.
We introduce mapquik, a novel strategy that creates accurate longer seeds by anchoring alignments through matches of k consecutively sampled minimizers (k-min-mers) and only indexing k-min-mers that occur once in the reference genome, thereby unlocking ultrafast mapping while retaining high sensitivity. We
show that mapquik significantly accelerates the seeding and chaining steps—fundamental bottlenecks to read mapping—for both
the human and maize genomes with > 96% sensitivity and near-perfect specificity. On the human genome, for both real and simulated
reads, mapquik achieves a 37 × speedup over the state-of-the-art tool minimap2, and on the maize genome, mapquik achieves
a 410 × speedup over minimap2, making mapquik the fastest mapper to date. These accelerations are enabled from not only minimizer-space
seeding but also a novel heuristic 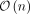 pseudochaining algorithm, which improves upon the long-standing
pseudochaining algorithm, which improves upon the long-standing 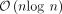 bound. Minimizer-space computation builds the foundation for achieving real-time analysis of long-read sequencing data.
bound. Minimizer-space computation builds the foundation for achieving real-time analysis of long-read sequencing data.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277679.123.
-
Freely available online through the Genome Research Open Access option.
- Received January 8, 2023.
- Accepted June 26, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








