

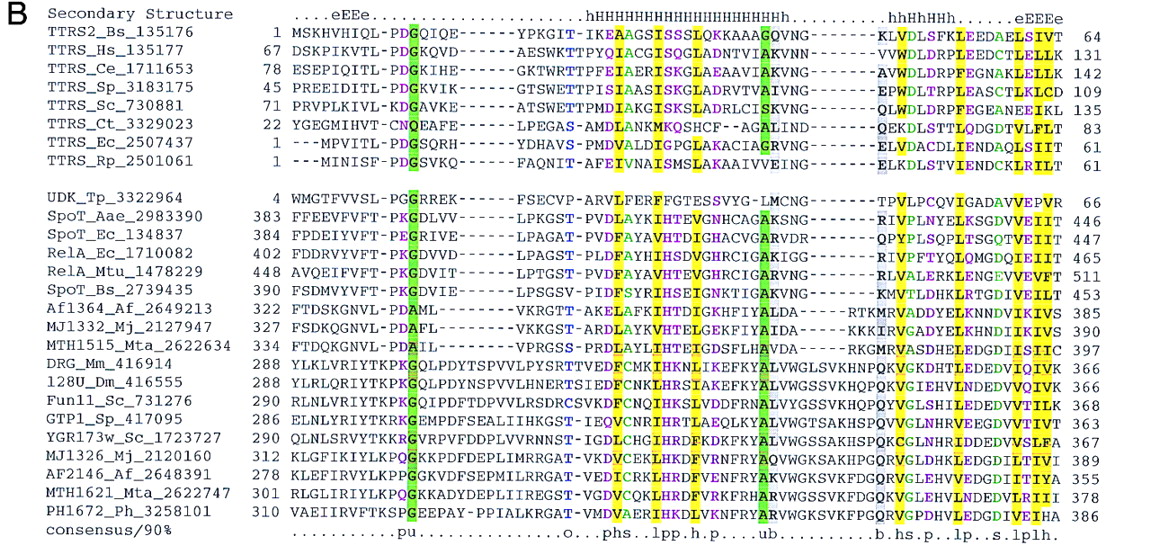


Previously undetected domain conservation in aaRS and proteins of other functions. (A) The GAD domain in bacterial AspRS and archaeal glutamyl-tRNA amidotrnasferases (GatB). (Top block of sequences) Archael GatB; (bottom block) bacterial AspRS. (B) The TGS domain in ThrRS, guanosine polyphosphatases (SpoT), OBG family GTPases, and uridine kinase (UDK) from Treponema. (Top block of sequences) ThrRS; (bottom block) other proteins containing the TGS domain. (C) The inactivated HD hydrolase domain in bacterial GlyRS β-subunit. (Top block of sequences) A selected subset of the HD superfamily hydrolases; (bottom block) β-subunits of bacterial GlyRS. (D) The winged helix–turn–helix domain in PheRS α-subunits from eukaryotes, archaea, and spirochaetes. (Top block of sequences) PheRS; (bottom block) a selected subset of other proteins containing the winged-HTH domain. The alignments were constructed on the basis of the PSI-BLAST results using the ClustalW program. The inclusion of each sequence in the alignments was statistically supported by PSI-BLAST results, with an e-value of at least 0.01. The left column includes the protein (gene) names, the abbreviated species name, and the gene identification (GI) nos. (following the underscore). A consensus derived using a 90% or an 85% cutoff is shown underneath the alignment, and the respective alignment columns are highlighted; (b) a big residue (E,K,R,I,L,M,F,Y,W); (h) a hydrophobic residue (A,C,F,I,L,M,V,W,Y); (s) a small residue (A,C,S,T,D,N,V,G,P); (u) a tiny residue (G,A,S); (o) a hydroxy residue (S,T); (p) a polar residue (D,E,H,K,N,Q,R,S,T); (c) a charged residue (K,R,D,E,H); (+) a positively charged residue (K,R,H). InA,B, and D, the numbers indicate the positions of the first and last residue of the aligned region in the respective protein sequence. The alignment in C consists of conserved blocks separated by variable spacers; the lengths of the spacers and the distances between the protein termini and the aligned regions are indicated by numbers. The secondary structure elements predicted using the PHD program, with the multiple alignment as the input (Rost and Sander 1994), is shown above the alignment inA, B, and D. [E(e)] indicates extended conformation (β-strand); [H(h)] α-helix; uppercase letters indicate the predictions made with a high level of confidence. InC, the line above the alignment indicates the predicted catalytic residues of the HD superfamily hydrolases (Aravind and Koonin 1998) that are replaced in the β-subunits of GlyRS. (Aae) A. aeolicus; (Af) A. fulgidus; (At) A. thaliana; (Bb) B. burgdorferi; (Bs) B. subtilis; (Ce) C. elegans; (Ct) C. trachomatis; (Dm) D. melanogaster; (Ec) E. coli; (Hi) H. influenzae; (Hs) H. sapiens; (Mj) M. jannaschii; (Mta) M. thermoautotrophicum; (Mtu) M. tuberculosis; (Ph) P. horikoshii; (Sp) S. pombe; (Sso) S. solfataricus; (Ssp) Sy. sp.; (Tm) Th. maritima; (Tp) T. pallidum; (VV) vaccinia virus.








