
Genetic Studies in the Sleep Disorder Narcolepsy
Abstract
Narcolepsy is a chronic neurologic disorder characterized by excessive daytime sleepiness and abnormal manifestations of REM sleep including cataplexy, sleep paralysis, and hypnagogic hallucinations. Narcolepsy is both a significant medical problem and a unique disease model for the study of sleep. Research in human narcolepsy has led to the identification of specific HLA alleles (DQB1*0602 andDQA1*0102) that predispose to the disorder. This has suggested the possibility that narcolepsy may be an autoimmune disorder, a hypothesis that has not been confirmed to date. Genetic factors other than HLA are also likely to be involved. In a canine model of narcolepsy, the disorder is transmitted as a non-MHC single autosomal recessive trait with full penetrance (canarc-1). A tightly linked marker for canarc-1 has been identified, and positional cloning studies are under way to isolate canarc-1 from a newly developed canine genomic BAC library. The molecular cloning of this gene may lead to a better understanding of sleep mechanisms, as has been the case for circadian rhythms following the cloning of frq, per, and Clock.
Sleep consumes almost one-third of any human lifetime, yet its biological function remains unknown. Electrophysiological studies have shown that sleep is physiologically heterogeneous. Sleep onset is first characterized by light nonrapid eye movement (NREM) sleep (stage I and II), followed by deep NREM sleep or slow-wave sleep (stage III and IV) and finally rapid eye movement (REM) sleep. This sleep cycle is ∼90 min long and is repeated multiple times during nocturnal sleep. REM sleep, also called paradoxical sleep, is characterized by low-voltage fast electroencephalogram activity, increased brain metabolism, skeletal muscle atonia, rapid eye movements, and dreaming. Total sleep deprivation and/or REM sleep deprivation are both lethal in animals.
NREM and REM sleep are mainly regulated by circadian and homeostatic processes. Recent studies have suggested that across the animal kingdom, circadian rhythms are regulated by similar negative feedback loops involving the rhythmic expression of RNAs encoding proteins that act to shut off the genes encoding them (Hall 1995; Dunlap 1996;Rosbash et al. 1996; Young et al. 1996). From a genetic perspective, much less progress has been made in the noncircadian aspects of sleep regulation. This review demonstrates that a genetic approach to narcolepsy will in time provide a novel insight into the molecular basis of sleep control.
Narcolepsy, a Disorder of REM Sleep Regulation
Narcolepsy most often begins in the second decade of life but may be observed at the age of 5 or younger (Honda 1988). The cardinal symptom in narcolepsy is a persistent and disabling excessive daytime sleepiness. Sleep attacks are unpredictable, irresistible, and may lead to continuing activities in a semiconscious manner, a phenomenon referred to as automatic behavior. Naps are usually refreshing, but the restorative effect vanishes quickly.
Sleepiness is not sufficient to diagnose the disorder. Narcoleptic patients also experience symptoms that are secondary to abnormal transitions to REM sleep (Aldrich 1992; Bassetti and Aldrich 1996). The most important of these symptoms is cataplexy, a pathognomonic symptom for the disorder. In cataplexy, humor, laughter, or anger triggers sudden episodes of muscle weakness ranging from sagging of the jaw, slurred speech, buckling of the knees or transient head dropping, to total collapse to the floor (Aldrich 1992; Bassetti and Aldrich 1996). Patients typically remain conscious during the attack, which may last a few seconds or a few minutes. Reflexes are abolished during the attack, as they are during natural REM sleep atonia. Sleep paralysis, another manifestation of REM sleep atonia, is characterized by an inability to move and speak while falling asleep or upon awakening. Episodes last a few seconds to several minutes and can be very frightening. Hypnagogic hallucinations are vivid perceptual dream-like experiences (generally visual) occurring at sleep onset. Sleep paralysis and hypnagogic hallucinations occasionally occur in normal individuals under extreme circumstances of sleep deprivation or after a change in sleep schedule (Aldrich 1992; Bassetti and Aldrich 1996) and thus have little diagnostic value in isolation.
Nocturnal sleep polysomnography is conducted to exclude other possible causes of daytime sleepiness such as sleep apnea or periodic limb movements (Aldrich 1992). The Multiple Sleep Latency Test (MSLT) is also carried out to demonstrate daytime sleepiness objectively. In this test, patients are requested to take four or five naps at 2-hr intervals, during which time to sleep onset (sleep latency) is measured. Short sleep latencies under 5 min are usually observed in narcoleptic patients, together with abnormal REM sleep episodes, referred to as sleep-onset REM periods (SOREMPs). The combination of a history of cataplexy, short sleep latencies, and two or more SOREMPs during MSLT is diagnostic for narcolepsy (Bassetti and Aldrich 1996;Mignot 1996). Note that many naps consist only of NREM sleep suggesting that there is also a broader problem of impaired sleep–wake regulation, with indistinct boundaries between sleep and wakefulness in narcolepsy (Broughton et al. 1986; Bassetti and Aldrich 1996).
The disorder has a large psychosocial impact. Two-thirds of patients have fallen asleep while driving, and 78% suffer from reduced performance at work (Broughton et al. 1981). Depression occurs in up to 23% of cases (Roth 1980). Treatment is purely symptomatic and generally involves amphetamine-like stimulants for excessive daytime sleepiness and antidepressive treatment for cataplexy and other symptoms of abnormal REM sleep (Bassetti and Aldrich 1996; Nishino and Mignot 1997).
Familial and Genetic Aspects of Human Narcolepsy
Narcolepsy–cataplexy affects 0.02%–0.18% of the general population in various ethnic groups (Mignot 1998). A familial tendency for narcolepsy has long been recognized (Roth 1980). The familial risk of a first-degree relative is 0.9%–2.3% for narcolepsy–cataplexy, which is 10–40 times higher than the prevalence in the general population (Mignot 1998).
In a Finnish twin cohort study consisting of 13,888 monozygotic (MZ) and same-sexed dizygotic (DZ) twin pairs, three narcoleptic individuals were found and each of them was discordant DZ with a negative family history (Hublin et al. 1994). In the literature, 16 MZ pairs with at least one affected twin have been reported and five of these pairs were concordant for narcolepsy (Mignot 1998). Although narcolepsy is likely to have a genetic predisposition, the low rate of concordance in narcoleptic MZ twins indicates that environmental factors play an important role in the development of the disease.
HLA DQA1*0102 andDQB1*0602 Are Primary Susceptibility Factors for Narcolepsy
Narcolepsy was shown to be associated with the human leukocyte antigen (HLA) DR2 in the Japanese population (Honda et al. 1984; Juji et al. 1984). DR2 is observed in all Japanese patients versus 33% of Japanese controls (Juji et al. 1984; Matsuki et al. 1988a). A similar association is observed in Caucasians, with >85% versus 22% DR2 positivity (Langdon et al. 1984; Billiard et al. 1986; Rogers et al. 1997). Strikingly however, the DR2association is much lower in African–Americans (65%–67% in narcoleptic patients vs. 27%–38% in controls) (Neely et al. 1987;Matsuki et al. 1992; Rogers et al. 1997). Further studies have shown that HLA DQ alleles, located ∼80 kb from the DRregion, are more tightly associated with narcolepsy than HLADR subtypes. More than 90% of narcolepsy–cataplexy patients across all ethnic groups carry a specific allele of HLA DQB1, DQB1*0602 (Matsuki et al. 1992; Mignot et al. 1994); this allele is present in 12%–38% of the general population across many ethnic groups (Matsuki et al. 1992; Mignot et al. 1994; Lin et al. 1997).DQB1*0602 is associated almost exclusively with DR2in Japanese (Lin et al. 1997) and Caucasians (Begovich et al. 1992), whereas it is observed frequently in association with DR2, DR5, or other DR subtypes in African–Americans (Mignot et al. 1994, 1997a). The increased DR–DQ haplotypic diversity in African–Americans explains the low DR2 association observed in this population.
To further characterize the DQB1 region in narcoleptic subjects, novel polymorphic markers were isolated and characterized (Mignot et al. 1997a). The markers tested included six novel microsatellite markers (DQCAR, DQCARII, G51152, DQRIV, T16CAR, and G411624R). DQA1, a DQ gene whose product is known to pair with DQB1-encoding polypeptides to form the biologically active DQ heterodimer molecule, was also studied. The results obtained are summarized in Figure1. The association with narcolepsy decreases in theT16CAR–DQB2 region (Mignot et al. 1997a) and in the DRB1 region (Mignot et al. 1994, 1997b). The G411624R andT16CAR microsatellites are complex repeats with drastically different sizes, all of which are frequently observed in narcolepsy susceptibility haplotypes, a result suggesting crossovers in the region. In the DRB1 region, association with narcolepsy is still tight with DRB1*1501 (DR2) in Caucasians and Asians but is significantly lower in African–Americans, which suggests crossovers in the region among ethnic groups.
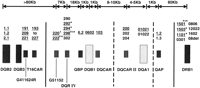
Schematic summary of the narcolepsy susceptibility region within the HLA complex. Genes and markers are depicted by vertical bars, alleles observed in narcoleptic patients are listed above each marker.DQB2, DQB3, DQB1, DQA1, and DRB1 are HLA genes and pseudogenes. QBP and QAP are the promoter regions ofDQB1 and DQA1, respectively. G411624R, T16CAR, G51152, DQCAR, and DQCARII are microsatellite CA repeats identified in the HLA DQ region (Mignot et al. 1997a).DQRIV is a compound tandem repeat of 4- and 2-bp units located between DQB1 and G51152. The DQA1*0102allele is subdivided into 01021 and 01022 based on a codon 109 synonymous substitution. Genomic segments in which frequent recombination was detected are indicated by vertical solid lines. Broken lines indicate rare possible ancestral crossovers detected in the area. Crossovers between T16CAR and G51152 occur within ethnic groups; crossovers between QAP and DRB1are frequently observed among ethnic groups (Mignot et al. 1997a). Note that the genomic region shared by most narcoleptic patients extends from a region between T16CAR and G51152 to a region between QAP and DRB1. No other genes were found in 86 kb of genomic sequence surrounding the DQB1*0602 gene (Ellis et al. 1997). Additional diversity is also found at the level ofG51152 and DQRIV, this being most likely due to a slippage mechanism rather than crossover (Lin et al. 1997; Mignot et al. 1997a). (+, Δ, *) Frequent alleles found predominantly in Caucasian, Asian, and African–American populations, respectively; (kb) kilobase pairs. Alleles frequently observed in theDQB1*0602/DQA1*0121 haplotype are underlined. DRB1*1501, DRB1*1503, and DRB1*1602 are DR2 subtypes.DRB1*1101 and DRB1*12022 are DR5 subtypes.
The DQA1*0102/DQB1*0602 haplotype is common in narcoleptic patients (Mignot et al. 1994). Other haplotypes with DQA1*0102but not DQB1*0602, such as DQA1*0102 andDQB1*0604, are frequent in control populations in all ethnic groups and do not predispose to narcolepsy. DQA1*0102 alone is thus not likely to confer susceptibility but may be involved in addition to DQB1*0602 for the development of narcolepsy (Mignot et al. 1994, 1997a).
Microsatellite analysis in the HLA DQ region revealed that only the area surrounding the coding regions of DQB1 andDQA1 is well conserved across all susceptibility haplotypes. Polymorphism can be observed in microsatellite and/or in the promoter regions flanking the DQB1*0602 and DQA1*0102alleles and in the region between these two genes (Mignot et al. 1997a). Mutations by slippage for some loci, and rare ancestral crossovers in a few instances, contribute to this diversity (Mignot et al. 1997a). Sequence analysis of DQ genes from narcoleptic and control individuals has revealed no sequence variation that correlates with the disease (Lock et al. 1988; Uryu et al. 1989; Ellis et al. 1997; Mignot et al. 1997a). No new gene was found in 86 kb of genomic sequence surrounding the HLA DQ gene (Ellis et al. 1997). A study on the dosage effect of DQB1*0602 allele on narcolepsy susceptibility revealed that DQB1*0602 homozygous subjects are at two to four times greater risk than heterozygous subjects for developing narcolepsy (Pelin et al. 1998). Taken together, these results strongly suggest that the DQA1*0102 andDQB1*0602 alleles themselves rather than an unknown gene in the region are the actual susceptibility genes for narcolepsy.
HLA DQB1*0602 Is Neither Sufficient nor Necessary for the Development of Narcolepsy
Of the general population, 12%–38% carry HLADQB1*0602, yet narcolepsy affects only 0.02%–0.18% of the general population. No sequence variation that correlates with the disease was detected in sequence analysis of DQ genes. Nevertheless, a few narcoleptic patients with cataplexy do not carry the DQB1*0602 allele (Mignot et al. 1992, 1997a). HLADQB1*0602 is thus neither necessary nor sufficient for development of narcolepsy–cataplexy.
Familial narcolepsy cases are more frequentlyDQB1*0602-negative than sporadic cases. Up to one-third of multiplex families may be DQB1*0602-negative (Mignot et al. 1996) and often display significant familial aggregation with no apparent linkage to HLA (Guilleminault et al. 1989; Mignot 1998). It is also noteworthy of that two of the five reported concordant MZ twin pairs are DQB1*0602-negative (Mignot 1998). Highly penetrant non-HLA susceptibility genes may thus account for most of the increased susceptibility observed in first-degree relatives of narcoleptics (Mignot 1998).
Canine Narcolepsy as a Model for the Human Disorder
Canine narcolepsy (Fig. 2) was first reported in a dachshund byKnecht et al. (1973) and in a poodle by Mitler et al. (1974). After a national search, narcolepsy was identified in numerous canine breeds, including Doberman pinschers, Labrador retrievers, miniature poodles, dachshunds, beagles, and Saint Bernards. All animals display similar symptoms, but the age of onset, severity, and the clinical course vary significantly among breeds (Baker et al. 1982). Several animals were donated and bred at Stanford University. The breeding of unrelated narcoleptic poodles and beagles (including some backcross matings) did not yield narcoleptic animals (Baker et al. 1982). Litters with several affected animals were discovered in Doberman pinschers and Labrador retrievers and used to establish a narcoleptic dog colony at Stanford (Baker et al. 1982). Genetic transmission in these two breeds is now well established as autosomal recessive with full penetrance, and the gene is designatedcanarc-1 (Baker et al. 1982; Mignot et al. 1991; Nishino and Mignot 1997). The same gene seems to be involved in both breeds, as Labrador–Doberman F1 hybrids also develop narcolepsy.

Narcoleptic Doberman pinschers in the midst of catapleptic attacks. The attacks were elicited by the emotion of playing together. Note that eyes are open. Cataplectic attacks involved an abrupt flaccid paralysis of all postural muscles.
Twenty years of experiments have demonstrated the validity of this model for narcolepsy research (Nishino and Mignot 1997, 1998). Similar to human narcoleptic patients, animals affected with the disorder display emotionally triggered cataplexy, fragmented sleep, and increased daytime sleepiness. Sleep paralysis and hypnagogic hallucinations cannot be documented because of difficulties in assessing the symptoms in canines. The validity of this model of narcolepsy has also been established through neurophysiological and neuropharmacological similarities with the human disorder. Pharmacological and neurochemical studies suggest abnormal monoaminergic and cholinergic mechanisms in narcolepsy both in human and canines (Aldrich 1991; Nishino and Mignot 1997, 1998). Interestingly, it is also possible to induce brief episodes of cataplexy in otherwise asymptomatic canarc-1 heterozygous animals using specific drug combinations (Mignot et al. 1993).
Is Narcolepsy an Autoimmune Disorder?
HLA class II molecules (DR, DQ, and DP) play a pivotal role in the genetic control of normal and pathological immune responses. These glycoproteins are constitutionally expressed on the surface of macrophages, monocytes, and lymphocytes, which present processed antigens to antigen-specific T lymphocytes, leading to their specific activation and proliferation. Autoimmune diseases such as rheumatoid arthritis, insulin-dependent diabetes mellitus, celiac disease, and multiple sclerosis are HLA class II-associated disorders (Charron 1990). In many of these diseases, the role of a specific HLA DR and DQ heterodimer is well documented in disease predisposition. A similar autoimmune mechanism was thus suggested for narcolepsy (Parkes et al. 1986), but all attempts have failed to substantiate this hypothesis (Rubin et al. 1988; Carlander et al. 1993; Mignot et al. 1995a). As a general rule, HLA class II-associated autoimmune diseases show a female preponderance, but no sex difference is observed in narcolepsy (Bassetti and Aldrich 1996). Systemic measurements of immune function in narcoleptics have also been found to be in the normal range. Lymphocyte subpopulations, erythrocyte sedimentation ratio, complement levels, and C-reactive protein are all within the normal range even in very early stages of the disease (Matsuki et al. 1988b). Rheumatoid factor, anti-nuclear antibodies, and all other attempts at demonstrating systemic autoimmune abnormalities have shown negative results (Rubin et al. 1988). Similarly, cerebrospinal fluid analysis also did not show increased frequency of oligoclonal IgG bands (Fredrikson et al. 1990).
The canine model of narcolepsy was also used to search for possible immune abnormalities. Histological analysis of the CNS of narcoleptic canines has shown no sign of localized inflammation or lymphocyte accumulation but did identify increased canine major histocompatibility complex (MHC) class II expression in the white matter around the time of disease onset (Tafti et al. 1996). Although a number of autoimmune disease pathologies can be transmitted or improved by bone marrow transplantation (van Bekkum 1993), bone marrow transplantation in two 8-month-old canine littermates with and without narcolepsy did not induce disease susceptibility or resistance (Mignot et al. 1995b). These results suggest that narcolepsy is not a classical autoimmune disease but do not exclude a short lasting or anatomically restricted autoimmune phenomenon.
Positional Cloning Studies in Canine Narcolepsy
Linkage studies using the canine model were initiated in the late 1980s. Linkage between canarc-1 and the dog MHC was excluded (Dean et al. 1989; Mignot et al. 1991). Screening of genetic markers, including minisatellite probes and functional candidate gene probes, revealed that canarc-1 cosegregates with a homolog of the switch region of the dog immunoglobulin μ heavy-chain gene (Sμ) (Mignot et al. 1991). The genuineSμ segments are involved in a complex somatic recombination process, allowing individual B cells to switch immunoglobulin class upon activation (Esser and Radbruch 1990). TheSμ-like narcolepsy marker has displayed a perfect cosegregation with the canarc-1 locus in 57 dogs (current lod score = 17.2 at 0% recombination; Faraco et al. 1998). Sequence analysis of the Sμ-like marker indicates that it has high homology to the true gene but is not a functional part of immunoglobulin switch machinery (Faraco et al. 1998). Chromosomal fluorescence in situ hybridization (FISH) has demonstrated that this marker is on a different chromosome than the veritable Sμ gene and the heavy chain immunoglobulin machinery.
Positional cloning efforts have been hampered by the lack of available cloning reagents in canines. A bacterial artificial chromosome (BAC) library that contains large canine genomic clones (average insert size of 155 kb and an eightfold coverage of the canine genome) was developed in collaboration with Roswell Park Cancer Institute to help our project and the canine genome community (H.R. Li, H. Kadotani, J. Faraco, L. Hinton, X. Lin, B. Zhao, K. Osoegawa, E. Mignot, and P.J. de Jong, unpubl.). With this library in hand, our laboratory has created a BAC clone contig greater than 1 Mb in the vicinity of canarc-1,which is under study (H.R. Li, H. Kadotani, J. Faraco, L. Hinton, X. Lin, B. Zhao, K. Osoegawa, E. Mignot, and P.J. de Jong, unpubl.). The BAC clones are used to isolate and test polymorphic microsatellite markers in the region. Once the susceptibility region is flanked, gene isolation and candidate gene studies will proceed in both canines and in the corresponding human region of conserved synteny.
Conclusion and Speculation
Narcolepsy is both a significant medical problem and a unique disease model. Research in humans has led to the identification of specific HLA alleles that predispose to the disorder. This has suggested the possibility that narcolepsy may be an autoimmune disorder, a hypothesis that has not been confirmed to date. Cells of the central and peripheral nervous systems and immune systems are known to interact at multiple levels (Morganti-Kossmann et al. 1992; Wilder 1995). For example, peripheral immunity is modulated by the brain via autonomic or neuroendocrinal interactions, whereas the immune system affects the nervous system through the release of cytokines. Cytokines have been shown to modulate sleep directly and have established effects on neurotransmission and neuronal differentiation (Krueger and Karnovsky 1995; Mehler and Kessler 1997). It is therefore possible that neuroimmune interactions that are not autoimmune in nature might be involved in the pathophysiology of narcolepsy.
NREM and REM sleep are mainly regulated by circadian and homeostatic processes. Single gene circadian mutations have been isolated from species as diverse as Arabidopsis (toc1),Neurospora (frq), Drosophila (perand tim), and mouse (Clock) (Hall 1995). Theper and Clock genes isolated in Drosophiliaand mouse, respectively, have been shown to belong to the same family, the PAS domain family (Hall 1995; Rosbash et al. 1996; Young et al. 1996; King et al. 1997). Analysis of frq, tim, andper demonstrate that circadian rhythms of diverse species are regulated by similar negative feedback loops in which gene products negatively regulate their own transcripts (Hall 1995; Dunlap 1996;Rosbash et al. 1996; Young et al. 1996). Putative homologs of theper gene have also been isolated in mammals (Albrecht et al. 1997; Tei et al. 1997). In mouse, RNAs for two per homologs are expressed rhythmically within the suprachiasmatic nucleus (SCN), a brain region with an established role in generating mammalian circadian rhythms (Shearman et al. 1997; Shigeyoshi et al. 1997; Tei et al. 1997).
Much less progress has been made in the noncircadian aspect of sleep regulation. Sleep can only be recognized and characterized electrophysiologically in mammals and birds, and single gene mutants for this behavior have not been described in the mouse. Canine narcolepsy is the only known single gene mutation affecting sleep state organization as opposed to circadian control of behavior. The molecular cloning of this gene may lead to a better understanding of the molecular basis and biological role of sleep, as has been the case for circadian rhythms following the cloning of frq, per, and Clock.
Acknowledgments
We thank P.J. de Jong for his invaluable help in the building of the canine BAC library, S. Hoshino for his work on the DQRIVrepeat, L. Hinton, X. Lin, H.R. Li, L. Lin, and A. Voros for assistance, and S. Nishino for his comments on the manuscript. Research has been funded by National Institutes of Health grants NS 23724 and NS/MH 33797 to E.M. and by a grant from the National Sleep Foundation. H.K. is supported by Sankyo Foundation of Life Science.
Footnotes
-
↵2 Corresponding author.
-
E-MAIL mignot{at}leland.stanford.edu; FAX (650) 498-7761.
- Cold Spring Harbor Laboratory Press








