
Sequence Assembly with CAFTOOLS
- Simon Dear1,
- Richard Durbin1,
- LaDeana Hillier2,
- Gabor Marth2,
- Jean Thierry-Mieg3 and
- Richard Mott1,4,5
- 1Sanger Centre, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, UK; 2Genome Sequencing Center, Washington University, St. Louis, Missouri 63108 USA; 3CRBM du Centre National de la Recherche Scientifique (CNRS), Route de Mende, Montpellier, France; 4SmithKline Beecham Pharmaceuticals, New Frontiers Science Park (North), Harlow, Essex, CM19 5AW, UK
Abstract
Large-scale genomic sequencing requires a software infrastructure to support and integrate applications that are not directly compatible. We describe a suite of software tools built around the Common Assembly Format (CAF), a comprehensive representation of a sequence assembly as a text file. These tools form the backbone of sequencing informatics at the Sanger Centre and the Genome Sequencing Center. The CAF format is intentionally flexible, and our Perl and C libraries, which parse and manipulate it, provide powerful tools for creating new applications as well as wrappers to incorporate other software. The tools are available free by anonymous FTP from ftp://ftp.sanger.ac.uk/pub/badger/.
Genomic sequencing is now a semi-industrial process that is being increasingly automated. The amount of finished sequence produced in large centers worldwide more than doubles each year. This effort has required a huge investment in bioinformatics, and new software is under continual development both within these centers and in the wider academic community. High-throughput sequence assembly is a complicated multistep pipeline, using many pieces of software, and we as users want to be in a position to use the best set of software tools, even if this causes problems reconciling the various data formats they use. In addition, because more than one tool may be suitable for the same task (e.g., for manually editing sequence assemblies) we also want to offer alternatives within the same framework.
Consequently we require a system that is flexible enough for us to evaluate and incorporate new software as it emerges and yet is easy to maintain and use. We have not found any existing product that meets these requirements completely, so our solution to this problem was to create the Common Assembly Format (CAF), a complete textual description of a sequence assembly, together with Perl and C libraries for parsing and manipulating CAF files, and applications written with these libraries to perform tasks for which no third-party software exists.
The purpose of this paper is threefold: (1) to describe CAF and its associated software package CAFTOOLS; (2) to illustrate their use for genomic sequencing at the Sanger Centre; and (3) to propose CAF as a standard format for developers and sequencing centers.
The CAF
Overview
CAF is a restriction of the data file format (.ace file format) conforming to a specific acedb(http://www.sanger.ac.uk/Software/Acedb/) schema for describing sequence assemblies. It is acedb-compliant, although using CAF does not require the use of acedb. A full acedb schema for CAF can be found at the official CAF web site,http://www.sanger.ac.uk/Software/CAF/. CAF is designed to be sufficiently comprehensive that any assembly engine/editor such asphrap (P. Green, pers. comm.), consed (Gordon et al. 1998), gap4 (Bonfield et al. 1995), acembly (J. Thierry-Mieg, unpubl.), FAKII (Larson et al. 1996; Myers 1996), and so forth, can derive all of the information it needs from the CAF file without reading any other data, except for trace information that is held in standard chromatogram format (SCF) files (Dear and Staden 1992). We have written tools to convert to and from each of these systems. Note that because CAF describes a superset of sequence attributes, passing an assembly through any of these editors may result in loss of information.
A sequence assembly is essentially a set of contigs, each contig being a multiple alignment of reads. In outline, the information we may need to store about each sequence is
- 1.
- The DNA sequence.
- 2.
- The base quality (a list of numbers indicating the confidence that the corresponding base in the sequence is correct).
- 3.
- The base positions (for reads only: a list of numbers indicating the location of the corresponding base within the SCF trace).
- 4.
- General properties (for reads only) such as the sequence template, name of corresponding trace file, etc.
- 5.
- Tags (regions of the sequence with some property, e.g., matching vector, repeat sequence, etc.).
- 6.
- Alignment of DNA onto the constituent reads, in the case of contigs, or of DNA onto original base calls in the case of reads.
CAF supports all of these features. In CAF the information associated with the sequence Name is divided into a maximum of four data types, which are represented as separate paragraphs of text, separated by blank lines, with header lines.DNA :Name 1. the DNA sequenceBaseQuality :Name 2. the base quality information for the DNABasePosition :Name 3. the base position information for the DNa, ie the trace coordinatesSequence :Name 4–6. all other information about the sequence
Throughout this paper, we will use Courier font for CAF reserved words and Italic Courier for CAF variables. The order of paragraphs within a CAF file is arbitrary. TheBaseQuality and BasePosition types are optional but the DNA and Sequence types are mandatory. Contigs and reads have the same DNA andBaseQuality types but specialized Sequencesubtypes. DNA sequence is represented as consecutive lines of text following DNA : Name. Base quality is represented as lines of space-separated integers followingBaseQuality : Name. The number of quality values must equal the length of the DNA. Base positions are represented similarly following BasePosition : Name.
The format of the data following aSequence : Name is variable. The minimum requirement is to specify the type (Is_Reador Is_Contig) and state (Padded or Unpadded, see the examples below). For clarity, we divide the Sequence data into simple and coordinate-sensitive attributes.
Simple Sequence Attributes
Sequence attributes that are not linked to coordinates have the general format
Attribute_type Value(s)
For example, the CAF fragment in Figure 1describes the sequence attributes of the readhh26e2.s1. Comments start with// and continue to the end of the line. New simple attributes can be created without constraint because CAFTOOLS will treat unrecognized attributes as text to be carried along unchanged with the associated Sequence object. The order of the attributes is arbitrary. Figure 1 details the most important and commonly used attributes, and a full list may be found at our web site.

An example showing how the attributes of the readhh26e2.s1 are described as a CAF Sequenceobject. The text following the // on each line are comments. This example illustrates most of the commonly used CAF read attributes.
Alignments and Coordinate-Sensitive Data
Those data that are coordinate dependent, for example, alignments and tags, are more structured because they must be parsed so that they can mirror changes to their associated DNAs. For example, editing, padding, or depadding a sequence alters the corresponding tag coordinates. The CAFTOOLS handle these changes transparently.
CAF stores two levels of alignment—that of the contig DNA to the read DNA, and that of the read DNA to the original base calls in the SCF trace. The alignment of a read onto a contig is represented by a series of statements of the form
Assembled_from Read c1 c2 r1 r2
which means coordinatesc1 to c2 in the contig align with r1 to r2 in the read. Coordinates start at 1. Ifc1 > c2 then the statement means align the reverse complement of r1 tor2 in the read. The lengths|c1–c2| and |r1–r2|must be the same. The alignment of a read to its original base calls in the SCF trace is similar:
Align_to_SCFr1 r2 t1 t2
that is, coordinatesr1 to r2 in the read correspond with t1 to t2 in the trace. Align_to_SCF is only applicable when the base positions of the DNA can be derived from the base position information held in the corresponding SCF trace files (recall that an SCF trace stores the trace data, the original base calls, and the mapping of each called base onto the trace). For certain purposes, for example, consed, we can override the SCF base calls and their positions by storing the coordinates explicitly in aBasePosition paragraph, in which case there is no need to use Align_to_SCF.
CAF supports padded and unpadded alignments. Padded means that gaps (–) have been inserted where required in the contigs and their aligned reads so that there is a one-to-one correspondence between the aligned sequences. In a padded assembly, there is exactly oneAssembled_ from line for each aligned read in a contig, and the DNA objects contain – padding characters.
In an unpadded alignment, all of the pads are removed from the DNA objects and there are multipleAssembled_from lines for each read in a contig. Within each Assembled_from line there is still a one-to-one correspondence between the read and contig.
Some applications, for example, auto-editor (described in Table1B) and gap4, require padded alignments. Others (e.g., applications that screen the DNA against known sequences like repeats) require unpadded. The programscaf_pad and caf_depadallow one to move transparently between padded and unpadded states, without losing information. In a padded alignment withBaseQuality information it is necessary to attach a quality value to each pad to keep the lengths equal. By convention this is interpolated from neighboring BaseQuality values.BasePosition data are treated similarly.
Summary of the CAFTOOLS
We illustrate these ideas with a hypothetical example. Suppose the alignment of Read_X to Contig_Y is
Read_X GCTGCCTTCGC–TTAAAA Contig_Y CAGCTGC-TTAGCGCTTAAAA
In padded coordinate space, positions [1,19] inRead_X align with [3,21] inContig_Y. However, the presence of pads inRead_X makes its alignment to its traceSCF_X complicated. Figure 2A shows a fragment of a CAF file describing the padded alignment. Note that the alignment of the read to the contig is attached to the contig’s sequence data and not the read’s. The equivalent unpadded description is shown in Figure 2B, where the coordi-nates now refer to the unpadded sequences. Note also that the alignment of the read DNA sequence onto the underlying base calls in the SCF file will change if insertions or deletions have been made to the read.

An example alignment in CAF, when the sequences are padded (A) and when they are unpadded (B).
The other major type of coordinate-sensitive data to consider is the tag. A tag is a region of a sequence with some property. The format must be one of
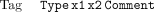
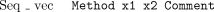
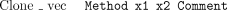
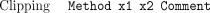 The Tag attribute means that positionsx1 to x2 of the sequence have property Type, optionally with the free textComment. This is used extensively to mark regions matching repeat sequences, or those that have been automatically edited with auto-editor. The special tagsSeq_vec, Clone_vec, andClipping are reserved for regions that match sequencing vector, cloning vector, or are high quality. These are held inside CAFTOOLS
as separate data structures. TheirMethod attribute is used to indicate which algorithm was used to generate the relevant coordinates (e.g., so that different quality
clip points can be represented).
The Tag attribute means that positionsx1 to x2 of the sequence have property Type, optionally with the free textComment. This is used extensively to mark regions matching repeat sequences, or those that have been automatically edited with auto-editor. The special tagsSeq_vec, Clone_vec, andClipping are reserved for regions that match sequencing vector, cloning vector, or are high quality. These are held inside CAFTOOLS
as separate data structures. TheirMethod attribute is used to indicate which algorithm was used to generate the relevant coordinates (e.g., so that different quality
clip points can be represented).
Finally, CAF supports the phrap concept of the “golden path” of a contig. This is a sequence of abutting intervals covering the contig’s DNA, such that each interval of the contig is associated with the read with locally the highest base quality. The format is a series of lines
Golden_path Read x1 x2
meaning Read provides the golden path for the interval[x1,x2]. Complete examples of CAF files can be found on our CAF web site.
The CAFTOOLS
We have written two libraries for reading, manipulating, and writing CAF files:
- 1.
- Perl-5 libraries, which are easy to use and are convenient for creating wrappers for software written by third parties. The general procedure is to extract the relevant data from the input CAF file, convert it into the format required by the program, run the application, parse the output back into CAF, and write an updated CAF file. For example, the CAF tagging applications are based on this model.
- 2.
- ANSI-C libraries, which are less flexible but are much faster and can handle very large data sets (up to 50,000 reads) without using too much memory. They also perform much more stringent data checking and understand which information is position-sensitive, so that coordinates are automatically changed in concert with DNA modifications. They also provide error checking that reports (a) references to nonexistent sequences, (b) sequence coordinates out of range (compared with length of DNA), (c) inconsistent alignments, and (d) mixed or unspecified pad states. They are suited for writing computationally intensive applications, such as the auto-editor, which requires access to trace data. Applications written with the C libraries can pad or depad the input data as required by a single function call. They can also handle multiple CAF databases simultaneously in different name spaces.
CAFTOOLS have been tested extensively under Digital Alpha OSF and Sun Solaris UNIX but should work on most UNIX platforms. They do not provide graphics. Most of the applications that we have written with the libraries act as UNIX-style filters, reading a CAF file on standard input, modifying it, and writing a new CAF on standard output. Command-line switches can modify the function of many applications. The only exceptions are those that split the CAF into multiple files (e.g., to prepare data for processing by phrap) or that merge multiple CAF files into one. This means it is possible to pipe together many processing modules in one command. For example, to auto-edit a gap4 database GAPDB.0 one could type
gap2caf -project GAPDB -version 0
-preserve | np_edit -scf | nd_edit
| caf2gap -project GAPDB -version 1
This will create a new edited database GAPDB.1. (The -preserveswitch ensures that the internal gap4 numbering of the sequences is retained. We use a special attribute,Staden_id, for this purpose.) In practice, we chop up the assembly pipeline at significant breakpoints and write intermediate temporary CAF files to disk so that we can switch modules more easily and perform a postmortem if an error occurs. Full details of how to run each application, including all command-line options, may be found on our web site. Table 1 summarizes the more important utilities and applications written using CAFTOOLS.
Most of the utilities run in under 10 elapsed sec on a cosmid-sized CAF file. The applications are slower, for example, the auto-editortakes 2–3 min to edit a cosmid (40 kb, ∼1000 reads). We can process a cosmid through our complete assembly pipeline in ∼15 min on a 433 Mhz DEC Alpha processor.
The Sequence Assembly Pipeline
CAFTOOLS are best illustrated by their use at the Sanger Centre and the Genome Sequencing Center. We use the following pipeline at the Sanger Centre for assembling reads from a bacterial clone (e.g., a cosmid or BAC) into contigs. Most genome sequencing centers that use shotgun sequencing follow a broadly similar work flow. Figure 3summarizes the pipeline and shows how CAF is integrated into it.
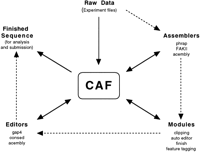
How CAF and CAFTOOLS are used in the sequence assembly pipeline. Broken arrows show the order in which data are processed; the solid arrows indicate actual data flow and how CAF is used as an interchange mechanism.
Preprocessing
As each sample comes off a sequencing machine, we preprocess it using asp (Automated Sequence Preprocessor) (M. Wendl, S. Dear, D. Hodgson, and L. Hillier, in prep.). asp is a chain of modules written in Perl, some of which call C programs, for example,phred (Ewing and Green 1998) andsvec_clip (Mott 1998). At the Sanger Centre,asp performs the following operations:
- 1.
- Query a central database to determine the world-unique name of the sample, the name of the parent clone (the “project”), the sequencing chemistry, the expected insert size, the sequencing vector, and the priming and cloning sites.
- 2.
- Base-call the sample using phred creating an SCF trace file and a file of phred base quality indices.
- 3.
- Determine quality clip points (i.e., the good-quality part of the read) from the phred base qualities. To do this we subtract 15 from each base quality index and then find left L and right R clip points such that the sum of adjusted quality values from L to R is a maximum.
- 4.
- Identify sequencing vector clip points by alignment of the original SCF trace to the expected vector sequence (svec_clip).
- 5.
- Screen the sequence against Escherichia coli and other possible contaminants.
Each sample processed by asp generates an Experiment file (Bonfield and Staden 1996) containing all the information generated about the sample in the run. A sample can be “failed” byasp if it has no high-quality region, is completely sequencing vector, or matches a contaminant. Samples that are passed byasp are moved, together with their associated trace files, into the relevant project directory for assembly. asp reports the fate (Pass or Failure, and the reason for failure) of each sample to a central database. The sequence assembly process will only start once the number of samples exceeds a threshold, typically ∼600 reads for a cosmid and ∼2000 for a PAC.
Assembly
The automated assembly process is controlled by a Perl script,phrap2gap. Each stage in the assembly pipeline is a module that accepts a CAF description of the current assembly, acts on it in some way, and writes out a new CAF file. Each CAF is a complete and consistent description of the current state of the assembly. Therefore, it is relatively easy to add or replace modules provided they conform to this pattern. For example, we have added support for the editorconsed, and for the sequence assembly engines FAKII andacembly. Figure 3 summarizes the pipeline. In greater detail, the steps are
- 1.
- Create a CAF file containing all of the raw data from individual Staden Experiment files (update_caf, exp2caf).
- 2.
- Extract from the raw CAF input files required by the assembly enginephrap (essentially a file of sequences and a file of base qualities). Assemble into contigs with phrap and merge back with the other information held in the raw CAF. The result is a new CAF file com pletely describing the reads and how they are assembled (caf_phrap, caf2phrap, cafmerge).
- 3.
- Clip back the assembled reads to their high-quality regions determined previously by asp (nd_clip). This process can occasionally create holes in contigs where no high-quality sequence occurs. phrap2gap offers the choice of splitting contigs (cafsplit) or re-extending reads back into their low quality parts to close the gaps (ne_clip).
- 4.
- Auto-edit the assembled reads, referring back to the original trace data (np_edit, nd_edit). Depending on the depth of coverage, up to 90% of the edits can be made at an error rate of less than one mistake per 50 kb.
- 5.
- Screen (using cafvector, cafalu, cafcgi, andcaftagfeature) the assembled contigs against various sequence data sets (e.g., cloning vector, known repeats, transposons) and tag any matching regions.
- 6.
- Analyze the assembly to choose directed reads for gap closure and to resolve ambiguities with finish (G. Marth and S. Dear, in prep.).
- 7.
- Convert the edited, tagged assembly into a gap4 database for manual finishing (caf2gap).
Further finishing reads are incorporated to close gaps in the contigs and strengthen the consensus in poor quality regions. To do this the user has the choice of adding the new sequences individually into the gap4 database and auto-editing again, or reassembling all of the data from scratch.
This pipeline uses the third-party applications phrap, cross_match (P. Green, pers. comm.), consed, blast (Altshul et al. 1990), and gap4 and two CAF applications auto-editor and finish, plus repeated use of several CAF utilities.
The Genome Sequencing Center uses a similar pipeline, except the auto-editing step is omitted and consed is used in place ofgap4. The Center also performs an internal quality-checking step on the final assembly (cafcop, pcop) before the sequence is submitted for analysis.
DISCUSSION
Historically, CAF dates from ∼1993 to 1994, growing out of our need to combine the assembly editor gap4 with the assembly engine phrap and other applications. At that time no existing interchange format was wholly appropriate for the task, for example, the Staden Experiment file format then did not represent contigs as distinct entities and relied on padded alignments. The early versions of CAFTOOLS were entirely Perl-based. We developed the C libraries initially to support auto-editing but soon recognized that the efficiency gains made developing a full C library worthwhile, particularly as we were starting to shotgun sequence 120-kb PAC and BAC clones.
CAF is not the only textual representation of sequence assemblies to be proposed. Staden Experiment file format has been suggested previously as a basis on which to form a standard interchange format (Bonfield and Staden 1996). The Boulder format (Stein et al. 1994) is used at the Whitehead Institute for purposes similar to CAF.
CAF may be viewed as a synthesis of many of the best features of other systems. Many of the CAF sequence attributes have exact correlates in Staden Experiment files, although the format is different and the description of multiple alignments byAssembled_from lines is similar to that used by acembly. We believe that CAF has the following advantages: (1) The plain text acedb file structure used by CAF means that files are easy to read and debug. (2) Holding all of the information about an assembly in a single file, rather than divided into individual sequence files makes it simpler to maintain consistent assemblies and to manipulate assemblies as single entities. (3) CAF is very flexible. We have been able to incorporate new software into our assembly pipeline relatively easily. (4) CAFTOOLS have been tested thoroughly and are in production use. Most of the total (to October 1997) 125 Mb of finished genomic DNA produced at our two sequencing centers was processed by some version of CAFTOOLS, and >50 Mbp was processed by the current version.
The Sanger Centre recently completed the 4.4-Mbp sequence ofMycobacterium tuberculosis(http://www.sanger.ac.uk/Projects/M_tuberculosis/) using CAFTOOLS applied to sequence data from a mixture of cosmids and a whole-genome library. The final stages of this project would have been much more difficult to complete without the infrastructure that CAF provided. This illustrates the power and importance of CAF.
The Sanger Centre is currently sequencing other pathogens, using whole-genome or chromosome libraries. We have found that, in general, assemblies covering more than 1 Mbp of genomic sequence are rather large to hold as single CAF files, although feasible on machines with large memories. More importantly, the final assembly must be split into pieces small enough to be completed by an individual while maintaining the integrity of the entire assembly. The natural solution is to store the CAF information in a database. It is possible to use acedbfor this purpose. Alternatively, one can use a relational database for CAF, and at the Sanger Centre we are currently implementing CAF in Oracle. The database communicates with other software by reading and writing CAF files, so that we can still use our existing CAFTOOLS while we benefit from being able to manipulate subsets of the data in a safe and efficient way. These developments have also led us to explore the extension of the CAF model to support partially assembled groups of contigs, to exploit relative contig order information from forward/reverse read pairs and restriction-digest information, although this is not yet part of the public distribution.
We anticipate that CAF will undergo further modification but that its basic principles will remain intact. We welcome suggestions for changes and additions to CAF. Our hope is that CAF will become more widely used for describing sequence assemblies and that developers will make use of it to the benefit of the wider sequencing community. As all workers in the field of bioinformatics recognize, an inordinate amount of time is spent interconverting file formats, to the detriment of actual software development. After >2 years of intensive development, testing, and production use, we believe CAF and CAFTOOLS are well-positioned to offer an effective solution to these problems in the domain of sequence assembly.
Acknowledgments
This work was supported by grants from the Wellcome Trust, CNRS, and the National Human Genome Research division of the National Institutes of Health. Also, we acknowledge the contributions of Rob Davies at the Sanger Centre, and Dave Hodgson, formerly at the Sanger Centre, to this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵5 Corresponding author.
-
E-MAIL richard_mott-1{at}sbphrd.com; FAX 44 1279 622200.
- Received December 1, 1997.
- Accepted January 29, 1998.
- Cold Spring Harbor Laboratory Press








