
Opportunities and Challenges Grow from ArabidopsisGenome Sequencing
A recent Cold Spring Harbor Laboratory meeting in December 1997 provided the first meeting on the Arabidopsisgenome featuring a unique combination of functional studies and sequencing efforts; it included a broad range of talks covering genome sequencing and analysis efforts, mapping and defining genes, and gene expression patterns and function. Significant points to come out of the meeting were that a number of international consortiums have completed substantial portions of sequence on all five chromosomes with 17 Mb of sequence currently available through various web pages and 8 Mb of annotated sequence available through GenBank. Although physical maps of three of the five chromosomes have not yet been completed, David Bouchez (INRA, Versailles, France) reported that >90% of the clones in the CIC (CNRS, INRA, CEPH) Arabidopsis YAC library have been anchored via hybridization to genetically mapped markers. This should greatly facilitate the construction of physical maps. Michael Mindrinos from the Ausubel laboratory (Massachusetts General Hospital, Boston, MA) reported the development of a new class of PCR-based marker, the SNAPs (single nucleotideamplified polymorphisms), which should greatly assist positional cloning efforts. Daphne Preuss (University of Chicago, IL) reported the use of tetrad analysis to place the centromeres on the genetic map (Fig. 1), taking advantage of the pollen mutant quartet1 (Preuss et al. 1994; Copenhaver et al. 1998). Interestingly, this analysis placed the centromeres very close to, but not necessarily within, the centromeric repeat blocks mapped recently by Round et al. (1997).
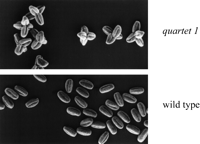
quartet1 mutant and wild-type pollen. The Arabidopsis quartet1 mutation results in tetrads remaining associated throughout pollen development. Crossing individual tetrads from recombinant inbreds allows tetrad analysis for centromere mapping. (Photo courtesy of Daphne Preuss, modified from Preuss et al. 1994.)
Sequencing Strides
Several technological developments are now being implemented to expedite complete sequencing of the genome. The SSP consortium has automated nearly all stages from clone selection through to sequence reactions. The widespread utility of these developments, in part or in total, may only be realized in the future when costs are reduced to allow sequencing of other plant genomes. It is the construction of sequence-ready BAC contigs that has largely facilitated rapid progress in sequencing; Francis Quetier (Genoscope–CNS, Evry, France), Nancy Federspiel (SPP consortium), and Steve Rounsley (TIGR, Rockville, MD) reported progress in sequencing the ends of as many as half of the BAC clones in two Arabidopsis libraries. Through this international collaboration, the sequential identification of overlapping BACs has become a routine procedure for many of the sequencing groups. An alternative approach, described by Marco Marra (Genome Sequencing Center, Washington University, St. Louis, MO), implements a new fingerprinting method (Marra et al. 1997) to the parallel assembly of sequence-ready BAC contigs inArabidopsis. Seventy-five percent of the available BACs have already been fingerprinted. Creators of software developed at the Sanger Centre (Cambridge, UK) have generated several large (2–3 Mb) contigs and promised to complete a BAC tiling path for most of the genome in the next few months. These contigs will both facilitate the final stages of genome sequencing and provide a powerful tool for positional cloning.
Sequence analysis has led to an accumulation of statistics on genome features such as gene density and gene structure. For example, the largest region of contiguous sequence obtained so far has been by ESSA, the European Union sequencing consortium. Mike Bevan (John Innes Institute, Norwich, UK) reported that the average gene density is one gene in 4.8 kb. Approximately 20% of genes belong to gene families and probably arise from gene duplication events followed by divergence. Interestingly, averages calculated for different chromosomal regions show minor variations. This may result from the clustering of specific gene types, each possibly having related expression patterns or involving common biological processes. One example of gene clustering may be found in the distribution of disease and defense response genes. Although many genes can be assigned a role based on functional characterization or sequence homology, ∼35%–40% of predicted genes have no significant database match or have homology only to hypothetical proteins in other organisms. Determining the function of these genes will be one of the major challenges of the future.
Dissecting Function
Although gene prediction programs are being optimized to enable accurate gene identification, another useful approach to this problem is comparative genomics. Thomas Mitchell-Olds (Max-Planck-Institute of Chemical Ecology, Jena, Germany) described the Arabis Genome Project involving a consortium focused on molecular and genetic characterization of wild relatives of Arabidopsis. Comparison of different gene expression patterns and molecular variation inArabis species will be used as an aid in defining gene function (Fig. 2). Although whole genome sequencing of an Arabis species is not currently under way, this is clearly a project for the future. Richard McCombie (CSHL) outlined early studies on cross-species genome sequence analysis and indicated some degree of conserved gene order between regions of theArabidopsis genome and genomes of the monocots maize and rice. This remarkable conservation will aid in obtaining gene sequences of the much larger monocot genomes and has the potential to define pathways of genome evolution.


Wild Arabis species potentially could be used to identify gene function based on outcrossed hybrids. (A) Arabis fecunda, a rare species endemic to western Montana with some ecotypes showing drought tolerance. (Photo by Jin-Zhong Lin.) (B) Halimolobos perplexa, a relative ofArabidopsis that grows in the northern Rocky Mountains. It is well adapted to hot, dry environments. (Photo by Thomas Mitchell-Olds.)
In an initial step toward addressing gene function, two main approaches, T-DNA insertion and transposon mutagenesis, have been used to generate populations of plants carrying random gene disruptions. These lines are being used to isolate plants carrying an insertion in a gene of interest. Typically, PCR using primers to the insertion element and to the gene of interest is carried out with pooled DNA samples. Subsequent deconvolution of pools is necessary to identify individual plants carrying the desired insertion. Steve Dellaporta (Yale University, New Haven, CT) and Andy Pereira (CPRO–DLO, Wageningen, The Netherlands) reported that, potentially, saturation coverage of the genome has been reached with multicopy T-DNA and transposon insertions, respectively. Much attention has been focused on T-DNA collections developed in France and the United States, as well as insertion lines carrying the maize transposable element Spm/En developed by a European consortium. It is not clear if pooling will be the most straightforward method for retrieving insertions. As an alternative, Rob Martienssen (CSHL) described the development of a collection of single-copy transposon insertions throughout the genome and has begun systematic sequencing of flanking DNA. The ultimate aim is to establish a whole genome or “core set” of gene disruptions that can subsequently be used for forward genetic analysis. Obviously, this approach is more ambitious than constructing pools of insertion lines, but the single-hit collection generated will be valuable for forward screens and will be searchable on the computer, rather than by reiterated PCR.
Sequencing the entire Arabidopsis genome will provide an invaluable tool for addressing the long-standing problem of genetic redundancy. Individual members within a gene family can each be targeted for mutagenesis. Systematic combination of individual mutations and analysis of the resulting phenotypes should define partners contributing to common biological functions, that is, genes that are fully redundant, as well as those that have overlapping functions. Patrick Krysan (University of Wisconsin, Madison) described such an approach in a study of the 14-3-3 gene family of which there are 10 members in Arabidopsis (Krysan et al. 1996). Plants carrying insertions in four of these genes do not have any noticeable phenotype. As a result these lines are being tested for more subtle phenotypes in a “gauntlet” of stress conditions similar to that used in the Saccharomyces Genome Deletion Project. However, unlike the yeast project, it is difficult to develop a method for multiplexing such phenotypic analyses. A novel approach to determine gene function is application of gene silencing as described by David Baulcombe (Sainsbury Laboratory, Norwich, UK). The silencing of host genes and homologous transgenes can be induced by local infection with viruses or infiltration with Agrobacteria each engineered to carry a region corresponding to the target gene. The silencing of gene expression spreads throughout the plant, occurring in tissues removed from the inoculation site (Fig. 3). The mechanism by which this occurs is as yet unknown (Palauqui et al. 1997; Voinnet and Baulcombe 1997). However, this method will be useful for studying the effects of disrupting expression of multigene families and genes essential for plant development.

Gene silencing using viral vectors. 35S green fluorescence protein is silenced after infection with antisense constructs. The silencing moves apically through the plant. (Photo courtesy of David Baulcombe.)
As the number of ORF and EST sequences has increased, it is increasingly feasible to investigate patterns of gene expression on a large scale. Multiple techniques for expression analysis were presented—each varying in cost, range of sensitivity, and ease of quantification. The prohibitive cost of microarrays produced by photolithography will make these genome chips out of reach for most plant biologists in the near future. On a more accessible scale, Mark Schena (Stanford University, CA) described microspotting and inkjetting techniques to obtain cDNA microarrays. Also, Doreen Ware and Randy Scholl of the Arabidopsis Biological Resource Center (Ohio State University, Columbus) described filter sets of the nonredundant EST collection to allow researchers to identify expression patterns of ESTs of interest. These technologies will have broad applications for identifying differentially expressed genes. Pieter Vos (Keygene, Wageningen, The Netherlands) outlined another approach to expression analysis whereby amplified fragment length polymorphism (AFLP) is applied to the analysis of cDNA populations. By increasing the specific bases on the primers, extremely low-level expression (1:106) can be detected and differences measured. Accurate quantification and identification of bands is the difficult aspect of this technology. Motoaki Seki (Shinozaki Laboratory, Riken, Tsukuba, Japan) has used bead-anchored YACs to enrich for cDNA sequences encoded by the YAC (Seki et al. 1997). Potential applications could be confirmation of ORFs and enrichment of coding sequence in plant species with large amounts of repetitive DNA.
Information Access
One of the many challenges to emerge from the cooperative effort to sequence the Arabidopsis genome is creating immediate, easy access to the information and resources generated. Both theArabidopsis thaliana Database (http://genome-www.stanford.edu/) and the Arabidopsis Genome Resource (http://synteny.nott.ac.uk/agr/agr.html) provide web sites from which most information can be retrieved, either directly or through links to other sites. One goal in the future will be to establish a unifiedArabidopsis genome database with fully annotated sequences linked to text and sequence search and viewing tools. Progress toward this end was described by Mike Cherry and David Flanders (Stanford University, CA), as well as Sean Walsh and Mary Anderson (University of Nottingham, UK). No less important will be easy access to other biological resources and information, such as tagged lines and their associated phenotypes, and global gene expression patterns. The clear impression from this meeting is that knowledge of theArabidopsis genome sequence is enabling the development of new and broad-based approaches that are likely to change the face of plant biology over the coming years.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL settles{at}cshl.org; FAX (516) 367-8369.
- Cold Spring Harbor Laboratory Press








