
Clusters of Resistance Genes in Plants Evolve by Divergent Selection and a Birth-and-Death Process
Abstract
Classical genetic and molecular data show that genes determining disease resistance in plants are frequently clustered in the genome. Genes for resistance (R genes) to diverse pathogens cloned from several species encode proteins that have motifs in common. These motifs indicate that R genes are part of signal-transduction systems. Most of these R genes encode a leucine-rich repeat (LRR) region. Sequences encoding putative solvent-exposed residues in this region are hypervariable and have elevated ratios of nonsynonymous to synonymous substitutions; this suggests that they have evolved to detect variation in pathogen-derived ligands. Generation of new resistance specificities previously had been thought to involve frequent unequal crossing-over and gene conversions. However, comparisons between resistance haplotypes reveal that orthologs are more similar than paralogs implying a low rate of sequence homogenization from unequal crossing-over and gene conversion. We propose a new model adapted and expanded from one proposed for the evolution of vertebrate major histocompatibility complex and immunoglobulin gene families. Our model emphasizes divergent selection acting on arrays of solvent-exposed residues in the LRR resulting in evolution of individual R genes within a haplotype. Intergenic unequal crossing-over and gene conversions are important but are not the primary mechanisms generating variation.
Plants, like animals, are continually challenged by a myriad of potential pathogens. There is increasing evidence that defense systems of plants are at least as complex as vertebrate defense systems. Unlike animals, however, plants do not have a circulatory system and therefore cannot rely on a specialized, proliferative immune system. Each plant cell has to be capable of defense, even though this defense is coordinated locally and systemically between cells. There are a variety of types of resistance genes and mechanisms, some induced and some constitutive (for review, see Godiard et al. 1994; Michelmore 1995; Hammond-Kosack and Jones 1997). Often, although not always, disease resistance in plants is determined by single, usually dominant, genes. The recent cloning of several such resistance genes (R genes) is providing insight into their function and evolution. The defense system of plants may be ancient and predate the evolution of the immune system; genes similar to plant R genes have been identified in mammals (Hammond-Kosack and Jones 1997; van der Biezen and Jones 1998).
In this review we consider what is known about the genomic organization and evolution of disease resistance genes in plants. The picture that is emerging for the organization and evolution of plant Rgenes is similar to that of the vertebrate major histocompatibility complex (MHC), T-cell receptor (TCR), and immunoglobulin genes. Therefore, although the specific types of genes involved are different, the evolutionary forces shaping the plant and vertebrate defense systems may be similar. We propose a model for the evolution of plantR genes that is adapted and expanded from a model developed for the evolution of vertebrate MHC and immunoglobulin gene families. Definitions of terminology can be found at the end of the text.
Classical Genetics
Resistance to many diseases, particularly those caused by biotrophic fungal diseases, is determined by individual members of families of dominant genes, each member conferring resistance to a specific strain of the pathogen. When parallel genetic studies have been made, specificity has often been shown to be conferred by a “gene-for-gene” interaction; for every gene for resistance in the host there is a matching gene for avirulence in the pathogen (Flor 1956). The gene-for-gene interaction has now been demonstrated or inferred in >20 diseases caused by taxonomically diverse pathogens and probably functions in many more (Flor 1971; Crute 1986). The gene-for-gene interaction is undoubtedly an oversimplification; however, it is a useful predictive genetic description of the interaction between plants and their pathogens.
Classical genetics has increasingly demonstrated that resistance genes tend to be clustered in the genome. Early studies were often limited to the study of a single pathogen. However, the advent of comprehensive genetic maps based on molecular markers has allowed genes for resistance to multiple diseases to be readily mapped relative to each other. R loci may be single genes with multiple alleles (Table1). The L locus in flax (13 alleles) andRPM1 in Arabidopsis (two alleles, presence or absence) are of this type. More commonly, R genes are organized in clusters that show varying levels of recombination between the component genes (Table 1). Genes within a single cluster can determine resistance to very different pathogens. On the basis of their clustered distribution and by inference from other cell–cell recognition systems, R genes were hypothesized to encode functionally and evolutionarily related members of recognition systems.
Clusters of Plant R Genes Identified by Classical Genetics
Molecular Evidence
Molecular data have supported this hypothesis. Nearly 20 Rgenes have now been cloned. Their isolation, structure, and potential function have been reviewed extensively (e.g., Staskawicz et al. 1995;Bent 1996; Baker et al. 1997; Hammond-Kosack and Jones 1997) and only features relevant to the evolution of R genes will be discussed here. All except two (Hm1 and mlo) seem to encode components of signal transduction systems. R genes encode a number of protein motifs in a variety of combinations (Fig. 1; Table 2) that are characteristic of receptors in yeast, Drosophila and vertebrates. The most prevalent class contains a nucleotide-binding site (NBS) and a leucine-rich repeat (LRR) region. Some are composed predominantly of an LRR region and transmembrane (TM) domain. Another is comprised of a LRR region and a protein kinase domain. Finally, one (Pto) is comprised of only a protein kinase domain but requires a NBS–LRR gene (Prf) for function. R genes therefore seem to encode receptors that detect the presence of the pathogen directly or indirectly. The signal passes through a network of signal-transduction cascades and a somewhat generic resistance response is elicited that may or may not involve host cell death (for review, see Dangl et al. 1996; Hammond-Kosack and Jones 1997).
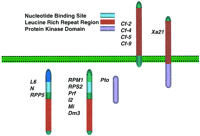
Major protein motifs shared between the deduced products of cloned resistance genes.
Clusters of Plant R Genes Characterized by Molecular Analysis
Restriction fragment length polymorphism (RFLP) analysis with most cloned resistance genes reveal clusters of related sequences, even when only a single resistance specificity had been detected by classical genetics (Table 2). L of flax is an interesting exception; the 13 specificities known at the L locus are truly allelic.M, the presumed homologous locus in this ancient tetraploid, has ∼86% nucleotide identity to L and comprises an array of ∼15 genes (paralogs); seven specificities are known at M(Ellis et al. 1997). Occasionally two functional sequences encode the same specificity, as in the case of Cf2 (Dixon et al. 1996). More usually only a single gene in the cluster determines resistance as shown by mutant or transgenic complementation analysis. The genetic and hybridization data therefore indicate that resistance genotypes should be considered as haplotypes rather than individual genes.
Molecular data is also revealing that clusters of R genes may contain sequences related in function but not sequence. Prf, an NBS–LRR gene, is within a cluster of five Pto homologs that encode protein kinases; both genes are required for resistance toPseudomonas syringae pv. tomato (Salmeron et al. 1996). Mi, an NBS–LRR gene, is linked loosely withCf2, a LRR–TM gene. Therefore, genetic linkage of phenotypically defined R genes does not necessarily imply sequence similarity.
There are a large number of sequences with similarity to Rgenes in plant genomes. PCR using degenerate primers designed to amplify sequences conserved between R genes has allowed identification of families of sequences from several plants (Kanazin et al. 1996; Leister et al. 1996; Yu et al. 1996; Lagudah et al. 1997;Leister et al. 1998). It is now facile to clone homologs but it remains difficult to prove individual specificities (Michelmore 1995). Large-scale sequencing of random expressed sequence tags (ESTs) and genomic clones has also identified numerous R gene homologs (Botella et al. 1997; Bevan et al. 1998). Extrapolating from current data, it seems likely that Arabidopsis has >400 resistance gene candidates (∼2% of its genes) and plants with larger genomes may have significantly greater numbers. Among the unanswered questions are: How many of these R gene candidates encode functional resistance genes? Is there a cost to expressing many R genes? What is the molecular basis of specificity? How do plants evolve new recognition specificities to keep pace with rapidly changing pathogen populations? What are the evolutionary forces shaping the size and structure of R gene clusters?
Basis of Specificity
The basis of specificity is unknown for most R genes.R-gene products are postulated to have receptor and effector domains (Fig. 2; Hammond-Kosack and Jones 1997) and patterns of nonsynonymous versus synonymous nucleotide-substitution ratios support this hypothesis (see below). A variety of motifs suggest involvement in protein–protein interactions; these could be involved in ligand binding or effector functions. Interactions between the putative components of a receptor complex have not been demonstrated yet. Mutations to losses of resistance are not particularly informative as to which domains determine specificity because mutations anywhere within the receptor or effector domains could abolish function. Various mutant alleles of Prf, RPS2, RPM1, andPto encode nonfunctional proteins that differ from wild type by a variety of single amino acids (Bent et al. 1994; Mindrinos et al. 1994; Grant et al. 1995; Salmeron et al. 1996; Scofield et al. 1996). Domain swaps between closely related proteins are likely to be much more instructive.
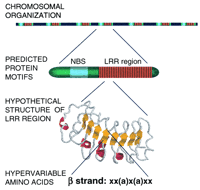
The organization and structure of resistance genes and their products. The color coding of the protein motifs is the same as for Figure 1. The hypothetical structure shown for a portion of the LRR region is prepared from a model for the thyrotropin receptor (Kajava et al. 1995). In the consensus sequence for the β-strand, (x) any amino acid; (a) aliphatic residues.
Pto is the best-characterized R gene. It is a small gene and diverse protein kinases have been extensively characterized structurally and biochemically. The product of the bacterial avirulence gene, AvrPto, interacts directly with the Pto gene product in the yeast two-hybrid system. The phenotypes of mutant and recombinant Pto genes in the yeast two-hybrid analysis correlate with biological activity in planta (Scofield et al. 1996;Tang et al. 1996). Domain swaps between Pto and the closely related paralog, Fen, identified a few amino acids in the ligand-binding domain (protein-kinase domain seven) as being critical for specificity (Scofield et al. 1996; J. Rathjen, J. Chang, D. Lavelle, B. Staskawicz, and R.W. Michelmore, unpubl.).
The LRR motif is a common motif that is thought to be involved in protein–protein interactions and known to be involved in ligand binding in porcine ribonuclease inhibitor (PRI; Kobe and Deisenhofer 1994, 1995). In PRI, the residues corresponding to the hypervariable residues in R gene products (see below) are part of a β-strand/β-turn structure of the LRR with a consensus sequence xxLxLxx. The conserved leucines (L) project into the hydrophobic core, whereas the other residues (x) form a solvent-exposed surface that is involved in ligand binding (Kobe and Deisenhofer 1995). The solvent-exposed surfaces of each LRR are arranged as a curved array; PRI accommodates its ligand by flexing along its backbone. InR genes, the conserved positions in the consensus sequence contain a variety of aliphatic residues (a; Fig. 2).R genes are unlikely to have as regular a structure as PRI because the amino acids in the backbone are more variable and there is less evidence that they form regular α-helices (Hammond-Kossack and Jones 1997; Jones and Jones 1997). Three-dimensional modeling suggests that the thyrotropin- and choriogonadotropin-receptor domains and LRRs of other proteins have less regular LRR structures but still comprise arrays of β-strand/β-turn structures (Kajava et al. 1995; Jiang et al. 1995; Kajava 1998).
Genetic rather than biochemical evidence currently provides the strongest evidence that the LRR interacts with a pathogen-derived ligand. Binding of an avirulence gene product or a plant protein to an LRR region has yet to be reported for any R gene product. Allelic comparisons and domain swaps between alleles of Lindicated that specificity was determined by the 3′ LRR-encoding part of the gene (Ellis et al. 1997). Sequences of the 5′ end of the LRR-encoding region of Cf genes are more variable than the 3′ regions (Parniske et al. 1997; Thomas et al. 1997). Detailed analysis of 11 Cf homologs revealed that in the first 16 LRRs, the amino acids in the xx(a)x(a)xx part of the LRR consensus were hypervariable, and the ratios of nonsynonymous to synonymous substitutions (KA:KS ratio) of the nucleotides encoding these residues (excluding the conserved aliphatic residues) were greater than one; therefore these residues may be under diversifying selection (Parniske et al. 1997). All types of LRR-encoding R genes exhibit a similar pattern (Table 3;Meyers et al. 1998b). The NBS region appears to be under purifying selection consistent with its proposed but unproven effector function. The LRR region, particularly the carboxy-terminal half of the LRR region encoded by Dm3 homologs in lettuce, contains an alternating pattern of hypervariable amino acids in the xx(a)x(a)xx consensus and intervening stretches of conserved residues that are predicted to be structural. The KA:KSratios for the nucleotides encoding these hypervariable residues are significantly greater than one, suggesting that there is an advantage to high amino acid diversity in this region. Therefore, both the amino-terminal end of the probably extracytoplasmic LRR regions of the LRR–TM type R gene products and the carboxy-terminal end of the predicted cytoplasmic LRR regions of the NBS–LRR type Rgene products show an alternating pattern of hypervaiable and conserved residues. The LRR region encoded by Xa21 and related paralogs show the same pattern. Regardless of the precise structure, the alternating pattern of variation and high KA:KSratios are evidence that these LRRs have a series of contact points for ligand binding (Fig. 2) that seem to be under diversifying selection.
Ratio of Nonsynonymous to Synonymous Nucleotide Substitutions (KA:KS) in Different Regions of Plant RGenes
A structure for LRR regions with arrays of potential ligand-binding surfaces has several implications for R-gene function. The most important implications are the extremely large number of binding specificities that could be encoded by groups of genes with such arrays (Fig. 2) and the ease with new binding specificities could be generated by recombination and gene conversion. In addition to different combinations of LRRs providing different binding characteristics, variation in amino acids in the backbone between the hypervariable regions might change the relative orientations of the β-strands providing another level of variation for binding specificity. The number of repeats does not seem to be highly conserved; this can vary from 14 to >40 (Jones and Jones 1997); Dm3 homologs encode at least 41 repeats (Meyers et al. 1998b). Cf2 differs from Cf5 by precisely six LRRs (Hammond-Kosack and Jones 1997). Different functional alleles of L vary in the length of their encoded LRR region (Ellis et al. 1997). Furthermore, LRR regions are large relative to predicted avirulence gene products. It is therefore unlikely that all LRRs within a particular protein are involved in binding each avirulence gene product. Different binding specificities could be generated by interactions with different LRRs within a protein, and a single protein could interact with a variety of ligands.RPM1 determines resistance against bacteria expressing two very different Avr genes (Grant et al. 1995). The tomatoMi gene confers resistance to both nematodes and aphids (Rossi et al. 1998). However, it has yet to be demonstrated in these cases whether the same or different molecules are detected by the plantR gene.
Post-transcriptional and post-translational modifications add further levels of potential complexity that have yet to be studied forR gene products. The prevalence and significance of the alternative mRNA splicing that seems to occur with N, L6, and RPP5 is unknown (Whitham et al. 1994;Lawrence et al. 1995; Parker et al. 1997). Amino acid differences between Cf gene products alter potential glycosylation sites (Jones and Jones 1997). For several mammalian extracellular receptors, the pattern of N-glycosylation influences ligand binding (Zhang et al. 1995).
Mechanisms Generating Variation in Specificity
Cycles of detection and mutation characterize the coevolution of plants and potential pathogens (Fig. 3). This is particularly evident with the specialized airborne pathogens, such as the mildews and rusts, giving rise to a boom-and-bust cycle (Suneson 1960) for the efficacy of resistance genes. However, the genetic events underlying the changes in the plant and pathogen are understood poorly.
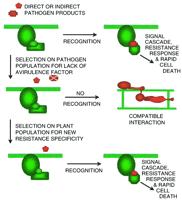
Alternating cycle of selection during coevolution of plant and pathogen.
Instability of some resistance loci, especially Rp1 of maize, led to the idea that clusters of resistance genes were inherently unstable, fast-evolving complexes. This instability is associated with recombination, and/or deletions (Sudupak et al. 1993; Anderson et al. 1996). Susceptible recombinants at Rp1 had both possible combinations of flanking alleles (Richter et al. 1995). Unequal crossing-over following meiotic mispairing and to a lesser extent gene conversion have been invoked as the major ways in which novel resistance specificities are generated (Pryor and Ellis 1993; Richter et al. 1995; Hammond-Kosack and Jones 1997; Hulbert 1997; Parniske et al. 1997). Such processes are almost certainly the primary mechanisms for concerted evolution of large multigene families such as vertebrate globins in which large amounts of homogeneous product are required (Scott et al. 1984). However, the roles of unequal crossing-over and gene conversion in the evolution of other multigene families are less clear and more controversial (Ohta 1991; Li 1997; Nei et al. 1997).
The assumption has been that resistance-gene complexes are dynamic, unstable, fast-evolving haplotypes and that high rates of unequal crossing-over and gene conversion are required to keep pace with changes in pathogen populations. However, high rates of unequal crossing-over and gene conversion would produce rapid divergence between haplotypes of different species. There would be high levels of polymorphism for both the number and sequence of R genes between haplotypes. Concerted evolution would tend to homogenize genes within a haplotype. There would be no obvious allelic relationship between genes in different haplotypes. Paralogs would be more similar than orthologs. In contrast to the above expectations, the recent comparisons of haplotype structure have been particularly informative and led to a very different picture. Studies on the Pto, Dm, and Cf clusters reveal an initially surprisingly stable picture with orthologs being more similar than paralogs.
The structures of three haplotypes of the Pto cluster in tomato have now been determined (D. Lavelle and R. Michelmore, unpubl.). One was the resistant haplotype that had been introgressed from Lycopersicon pimpernellifolium and two were susceptible haplotypes from Lycopersicon esculentum. Four to sixPto homologs are present within 65 kb along with a single copy of Prf. The closest relative of each gene is an ortholog rather than a paralog resulting in an obvious allelic relationship between homologs in different haplotypes. Only one pseudogene was apparent in the sequence of one haplotype as well as two partial gene segments. The cluster has evolved by a series of ancient duplications and more recent deletions. Unequal crossing-over with exchanges both between and within coding regions has occurred but has been infrequent.
The major cluster of resistance genes in lettuce is comprised of over 24 R gene candidates (RGCs) spanning several megabases (Meyers et al. 1998a). Genetic and mutation data indicate that oneRGC2 sequence encodes Dm3. The sequences within a genotype vary from 55%–95% amino acid identity. This is considerably greater than the differences observed between RGCs from different haplotypes and species (S.-S. Woo and R. Michelmore, unpubl.). The majority of RGCs sequenced appear to be functional and expressed, although some are clearly pseudogenes. The numbers of RGCs vary between genotypes. Several spontaneous losses of resistance have resulted from deletions but the involvement of RGCs at the breakpoints has not been demonstrated (D. Chin and R. Michelmore, unpubl.).
Three Cf4/9 haplotypes that originated from different tomato species have been sequenced (Parniske et al. 1997). The haplotype with no known resistance specificities contained only a singleR-gene homolog. The other two haplotypes spanned ∼35 kb and contained five paralogs, all oriented in the same direction. In all three haplotypes, the R-gene cluster was flanked by convergently oriented genes with similarity to plant lipoxygenases (Lox). In addition, the majority of the R-gene homologs within each cluster were flanked by partial Loxsequences indicating that Lox sequences had been duplicated along with the R gene. There were no other genes located within the cluster. Extensive blocks of sequence similarity were detected between the paralogs. The lengths and positions of these blocks varied greatly; however, their order was not changed indicating evolution by deletion following duplication of the R gene. There were no obvious R pseudogenes, which was in contrast to the large amount of intergenic rearrangements observed. Some homologs from different haplotypes had almost identical sequences indicating little sequence exchange between paralogs. Other homologs seemed to be a patchwork of short stretches of sequence similarity indicating the involvement of recombination or gene conversion.
Only a single haplotype of the Xa21 cluster in rice has been partially characterized at the sequence level (Song et al. 1997). This revealed two subfamilies of paralogs. However, the subfamilies had a mixture of sequences 5′ to the open reading frame (ORF) suggesting that there was a hot-spot for recombination just 3′ to the start codon and exchange of promoter regions between paralogs.
The sequencing of more clusters from multiple genotypes will reveal the relative orientations of genes, the numbers of pseudogenes, the degree of sequence divergence in intergenic regions, and the relationship between physical position and sequence similarity of the genes. This will provide additional data on the frequency of gene conversion tracts and insertions/deletions characteristic of unequal crossing-over events.
Birth-and-Death Model for Evolution of Resistance-Gene Clusters
A model is required that takes into account the probable structure of R-gene products and accommodates the observed stability of sequences between haplotypes as well as the genetic data on instability, yet allowing the rapid generation of new recognition specificities. Although unequal crossing-over is clearly occurring at a low frequency, it is not occurring at a high enough rate to homogenize sequences and it is probably too infrequent to generate new specificities to allow coevolution with pathogen populations. Frequent unequal crossing-over and gene conversion might actually be counterproductive to generating and maintaining variation. Therefore, we propose that NBS–LRR-type R genes are evolving mainly by divergent evolution of individual genes and a birth-and-death process (Fig. 4) similar to that envisaged for MHC and immunoglobulin genes (Nei et al. 1997).
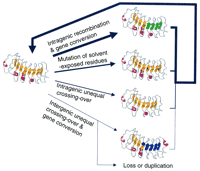
Consequences of genetic changes in resistance genes on the LRR region. Only a portion of the hypothetical structure of the LRR region is shown (see Fig. 2).
Salient features of the model include the following:
- 1.
- The majority of changes in specificity are caused by interallelic recombination and gene conversion that alter the combinations and/or orientations of the arrays of solvent-exposed residues in the LRR region. Recombination and gene conversion between paralogs is rare.
- 2.
- Further changes in specificity result either from mutations in the solvent-exposed region of the LRRs caused by random nucleotide changes or from interallelic unequal crossing-over or gene conversion because of mispairing within the LRR region.
- 3.
- Continued mutation and interallelic recombination will result in the selection of variants encoding increasingly effective resistance genes (increased ligand binding affinities?).
- 4.
- Sequences with advantageous ligand-binding characteristics will increase in the population. Multiple specificities are maintained in the population because of frequency-dependent selection and hitch-hiking caused by selection acting on the whole cluster of resistance genes.
- 5.
- Rare unequal crossing-over events result in duplications and deletions of either single genes or blocks of genes as well as occasional formation of chimeras between paralogs.
- 6.
- Recently duplicated sequences are unstable because of a high degree of sequence similarity resulting in relatively frequent unequal crossing-over. This leads to further duplications and deletions.
- 7.
- Rapid divergence of intergenic regions (and possibly introns) reduces the frequency of unequal crossing-over. Variants and derivatives become fixed in the haplotype.
- 8.
- Duplicated genes diverge (1 and 2 above). Some will have altered ligand-binding characteristics; others will become pseudogenes.
Evidence for Model and Implications
Clearly some components of the model remain unproven. However, the model is consistent with current data and does reconcile the maintenance of sequence diversity within haplotypes with the changes in specificity that are associated with recombination. It also explains how the proposed structure of the LRR allows different genetic rearrangements to generate a vast repertoire of resistance specificities (Fig. 4). The solvent-exposed amino acids in the LRR region may not be the only residues determining specificity; KA:KS analysis would not identify critical residues under selection for increased variability that are surrounded by conserved residues. However, the general aspects of the model are still valid regardless of which residues are the primary determinants of specificity.
1. Variation Is Generated Predominantly by Interallelic Recombination
The high lefvel of polymorphism in vertebrate MHC genes may be generated by several mechanisms. Interallelic recombination and gene conversion at the mammalian MHC class IB locus generates variation substantially faster than point substitutions (Watkins et al. 1992; Parham and Ohta 1996). Phylogenetic analysis of the MHC demonstrated that polymorphic alleles, as defined by their position within the cluster, almost always formed monophyletic groups; this suggests that there has been little gene conversion between paralogs (Nei et al. 1997). Closely related mammals all have MHC class IA, B, and C genes; orthologs are more similar than paralogs indicating that there has been little genetic exchange between paralogs.
The model proposes that the evolution of resistance in plants is occurring primarily at the single-gene level (Fig. 4). Interallelic recombination and gene conversion could generate considerable variation and explain much of the genetic data on the instability of resistance genes. If the LRR regions of resistance-gene products have arrays of solvent-exposed β-strand/β-turn structures, there are probably multiple ligand-binding points spread along the LRR region. Recombination between alleles would result in rapid generation of new ligand-binding properties without the need for high rates of nucleotide substitution. At least some of the instability at L andRp1 observed in heteroallelic crosses could be caused by interallelic recombination. The requirement for multiple binding points would result in interallelic recombinants that had lost specificity with either combination of flanking markers, as observed withRp1 (Richter et al. 1995; Hulbert 1997); there is no need to invoke intergenic unequal crossing-over or gene conversion.
Unequal crossing-over between members of a tandemly repeated multigene family is not a prerequisite for variation. There is only one copy of the gene at the L locus in flax, yet more specificities have been identified at L (13) than any of the resistance clusters (Islam and Shepherd 1991; Lawrence et al. 1995). Comparisons between alleles of L detected variation in both the number and sequence of the LRR at the carboxy-terminal end of the predicted protein (Ellis et al. 1995, 1997). The L2 allele has four copies of a 150-bp repeat rather than the two found in L6. Therefore, there can be significant variation without exchange between paralogs. Deletions within M and RPP5 that resulted in losses of resistance seemed to have been caused by intragenic recombination within the LRR-encoding region (Anderson et al. 1997;Parker et al. 1997).
The greater sequence divergence within than between haplotypes observed at the Pto cluster (D. Lavelle and R. Michelmore, unpubl.), the major cluster in lettuce (S.-S. Woo and R. Michelmore, unpubl.), and the Cf4/9 cluster (Parniske et al. 1997), indicates that unequal crossing-over and gene conversion between paralogs is not occurring frequently enough to homogenize the sequences within a haplotype. The more rapid divergence of Pto pseudogene sequences supports the same conclusion (D. Lavelle and R. Michelmore, unpubl.). Therefore the pattern of sequence divergence for multiple clusters of R genes is more consistent with a birth-and-death process than concerted evolution (Fig. 5).
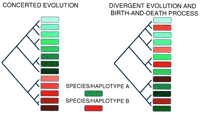
Phylogenetic distribution of sequence variation expected for homologs under different types of evolution.
2. Mutations in Individual Genes are an Ultimate Source of Novel Variation
A variety of types of mutation could lead to changes in specificity. The high KA:KS values and ratios for regions encoding the putative solvent-exposed residues in the LRRs ofCf4/9, Dm3, L, M, I2, Xa21, and their respective homologs all indicate the significance of nonsynonymous nucleotide substitutions in these regions (Table 3). Interallelic unequal crossing-over or gene conversion could change the number of LRRs. The repeated nature of the region implies that replication slippage (Hancock 1995) is also a possibility.
The relative importance of interallelic recombination and gene conversion (1) versus de novo changes (2) in the rapid and continuous generation of new resistance specificities is impossible to evaluate at present. Recombination and gene conversion seem to be the predominant mechanisms generating allelic variation at the MHC locus (Parham and Ohta 1996). However, point mutations accumulating over long periods of time seem to be the primary source of novel polymorphism in vertebrate MHC genes (Nei and Hughes 1991; Li 1997). Given that the LRR region is much larger than the binding region of the MHC protein, opportunities for interallelic recombination and gene conversion would seem to be correspondingly greater. The combination of multiple LRRS within each gene and the large number of R genes present in the genome provides numerous opportunities for advantageous mutations to arise. A few amino acid changes can have a large effect on specificity as shown by analysis of Pto, Fen, and related proteins (J. Rathjen, J Chang, D. Lavelle, B. Staskawicz, and R.W. Michelmore, unpubl.). Accumulation of amino acid changes over long periods of time has been proposed as the mechanism for evolution of gametophytic S-allele specificities (Tsai et al. 1992).
3. Resistance Specificities May Mature through Repeated Cycles of Selection
Evolution of new specificities may not be a one-step process. More likely, the initial interaction with a new ligand may be weak; there would then be selection for progressively tighter binding, if this results in more effective resistance. This is similar conceptually to somatic antibody maturation that occurs in T cells of vertebrates (Nossal 1992; Mueller and Jemmerson 1996). Variation in ligand binding is consistent with the variability of dominance observed forR-gene action and the variation in resistance phenotypes in different interactions (although there are several alternative explanations).
4. The Frequency of R Alleles Will Be Determined by the Interaction of a Variety of Forces
It seems intuitive that rare advantageous alleles of individual genes will tend to increase in the population until the frequency of virulent components of the pathogen population also increase to the point at which there is negligible selective advantage. In reality, however, the situation is likely to be much more complex. Selection will act on whole haplotypes. The rates of recombination across the cluster will influence the degree of independence of each paralog. Different paralogs within a haplotype will be under selection by the same or different pathogen populations and may confer different levels of fitness in different environments. The dynamics of individual haplotypes will become increasingly complex as the number of individual paralogs that are effective against pathogens at a particular location increases. This will be further complicated by the degree and stability of heterogeneity in the biotic and abiotic environments.
A variety of mechanisms have been proposed to explain the maintenance of polymorphism in vertebrate MHC and immunoglobulin genes. These include linkage disequilibria, cyclical selection, heterozygote advantage, and overdominant selection, as well as frequency-dependent selection caused by minority-dependent advantage (Nevo and Beiles 1992;Li 1997; Nei et al. 1997). Most of these selective forces are not mutually exclusive and it is still the subject of debate as to which of these mechanisms most influences the levels of MHC and immunoglobulin polymorphism. Very little is known of the population genetics ofR haplotypes and individual R genes within them. This will be a productive research area as the tools are now available to investigate the influence of different evolutionary forces on R genes.
5. Unequal Crossing-Over Is Significant
Clearly some unequal crossing-over is occurring. The copy number ofR-gene paralogs varies between haplotypes for Cf9, Pto, and Dm3 (Anderson et al. 1996; Parniske et al. 1997; D. Lavelle and R. Michelmore, unpubl.; D. Sicard, E. Nevo, and R. Michelmore, unpubl.). Sequencing of multiple homologs at theCf4/9 and Pto clusters indicated the occurrence of meiotic mispairing and recombination either within or between the coding regions for R genes. Two near-identical paralogs are present in the Cf2 haplotype, each of which can determine theCf2 specificity (Dixon et al. 1996). Sequence analysis ofDm3 paralogs suggested at least one chimeric gene (Meyers et al. 1998a). Spontaneous mutations of Dm3 and Cf9 are caused by deletions, although the involvement of recombination was not demonstrated (Anderson et al. 1996; Parniske et al. 1997). The instability at Rp1 in homoallelic crosses is associated with exchange of flanking markers and seems to be strong evidence for intra- or intergenic unequal crossing-over (Ritcher et al. 1995; Hulbert 1997). It will be interesting to confirm the underlying genetic events when this gene is cloned.
Unequal crossing-over and gene conversion are clearly important in the evolution of vertebrate MHC and immunoglobulin genes. However, although still subject to debate, these mechanisms are now not thought to be primarily responsible for generating new specificities. Instead phylogenetic analyses suggest that these families have evolved by a birth-and-death process (Nei et al. 1997). The numbers of MHC class I and II genes varies between haplotypes. Distantly related vertebrates have different sets of class IA genes, indicating that the haplotypes have evolved by duplication, divergence, and deletion of different paralogs. The human genome contains about 90 V H, 80V κ and 50 V λ genes encoding immunoglobulin components; however, the precise numbers vary between haplotypes. The numbers also vary dramatically between species. The phylogenetic trees are not correlated with the physical organization of the haplotypes; therefore each of the V H,V κ, and V λ clusters seem to have been generated by combinations of duplication, deletion, and translocation rather than repeated duplications alone (Nei et al. 1997).
The consequences of an unequal cross-over differ depending on whether the cross-over point is located in the noncoding, intergenic regions or within the coding regions of the paralogs. Both types of exchanges result in duplication and deletion of whole genes; however, only the former generates chimeric genes with potentially new specificities or expression patterns. Sequence comparisons of paralogs of Xa21demonstrate the shuffling of the 5′ region relative to the ORF (Song et al. 1997).
6. The Stability of Resistance Will Depend on the Similarity Between Haplotypes and the Mating System
In crosses between haplotypes of close but not identical structure, there will be the potential for extensive mispairing and unequal crossing-over because of structural hybridity. Cf4 andCf9 regions exhibit a fair amount of similarity. Cf9was stable when homozygous but unstable when heterozygous withCf4; the exchanges occurred between similar sequences in intergenic regions rather than within genes (Parniske et al. 1997). Extensive regions of hemizygosity will occur between individuals with divergent haplotypes. This will result in repression of recombination as has been observed in crosses involving resistance genes introgressed from wild species (Ganal and Tanksley 1996).
Consequences of the breeding system on the structure of R haplotypes have yet to be investigated. The clusters characterized so far at the molecular level are from inbreeding species predominantly. An inbreeding mating system will tend to favor the accumulation of structural variants because they would rapidly become homozygous; this would tend to promote instability in crosses with near relatives but repress recombination in more distant crosses. Outbreeding species, such as maize, may be more unstable as duplications would tend to be hemizygous, therefore promoting a variety of pairing possibilities. It will be interesting to compare the structure and variation ofRp1 haplotypes in corn to those characterized from rice, tomato, lettuce, and Arabidopsis, all inbreeding species. Significant instability was detected when Rp1 was homozygous and flanking markers were heterozygous. The underlying genetic events have yet to be characterized but may reflect a focusing of recombination to regions of homozygosity.
7. Rapid Divergence of Intergenic Regions Will Fix Variation
Sequencing of multiple haplotypes of the Cf4/9 region detected considerable variation in intergenic regions that appeared to have been generated by deletions. Most intergenic regions were distinct from each other. Sequencing of the Pto cluster revealed little sequence similarity 5′ and 3′ to paralogs. There were variable numbers of insertions/deletions between haplotypes. Structural variation in intergenic regions and introns would reduce meiotic mispairing (Xu et al. 1995; Dooner and Martinez-Ferez 1997) and therefore unequal crossing-over and gene conversion. Structural rearrangements restrict recombination at the S locus inBrassica species (Boyes et al. 1997). Once divergence exceeds the threshold for pairing, variants or derivatives will tend to become fixed in the haplotype.
8. Duplicated Genes Will Diverge
Duplication of a sequence relieves the selective pressure on all but one copy allowing divergence. Genes with new specificities as well as pseudogenes will be generated by interallelic recombination and mutation (1 and 2 above). Genes that evolve advantageous new specificities will tend to increase in the population (4 above). Pseudogenes will tend to be lost because of infrequent unequal crossing-over (5 above).
The relative frequencies of active genes and pseudogenes have yet to be determined for R homologs. Only one pseudogene was apparent from the sequence of two haplotypes of the Pto cluster (D. Lavelle and R. Michelmore, unpubl.). The majority of genes sequenced from one haplotype of the major cluster in lettuce had complete ORFs and were expressed; however, some genes were clearly pseudogenes (Meyers et al. 1998b). Dysfunctional genes caused by point mutations are quite common and interspersed with functional genes in the vertebrate MHC gene clusters (Trowsdale 1995). The humanV H region contains 50 functional and 40 nonfunctional genes.
The Role of Transposable Elements
Transposable elements of several types are major components of most plant genomes and clusters of resistance genes are no exception. Such elements could play several roles in the evolution of resistance genes. Insertion of the same element to two positions flanking a Rgene could provide the primary duplicated sequences allowing unequal crossing-over and the initial duplication of the R gene sequence (Wessler et al. 1995). Insertions will tend to increase misalignment and therefore increase the chances of unequal crossing-over when hemizygous. However, when homozygous, insertions will tend to decrease the chances of misalignment and therefore contribute to the divergence of intergenic regions. Several retrotransposon-related sequences are present in unique positions between Pto paralogs (D. Lavelle and R. Michelmore, unpubl.). Eleven different families of transposable elements were identified at the Xa21 cluster (Song et al. 1997). Footprints left by the excision of elements can result in insertion or deletion of amino acids. A revertant of L6 had three additional amino acids in the 5′ region of the gene (Ellis et al. 1997). Transposons also provide the opportunity for changing expression patterns by adding or disrupting regulatory elements.
The Role of Pseudogenes
Sequencing of paralogs of Xa21, Cf9, Dm3, and Pto has revealed the presence of pseudogenes (Parniske et al. 1997; Song et al. 1997; Meyers et al. 1998b; D. Lavelle and R. Michelmore, unpubl.). Pseudogenes tend to evolve much faster than functional genes (Ota and Nei 1994) and therefore represent the possibility of more rapid evolution of new specificities. However, longer branch lengths for pseudogenes in phylogenetic analyses of resistance genes again indicate that neither intergenic gene conversion or unequal crossing-over play a major role in homogenizing these genes (Ota and Nei 1994; D. Lavelle and R. Michelmore, unpubl.).
Given that a gene can become inactive because of mutations in regions other than those determining specificity and that recombination and gene conversion can shuffle sequences between alleles or paralogs, pseudogenes may be potential reservoirs of useful variation rather than just degenerate genes. Chicken has only one functionalV H gene and approximately 80 V Hpseudogenes in the genome. Antibody diversity is generated by somatic gene conversion between the functional gene and pseudogenes (Reynaud et al. 1989; Ota and Nei 1994). Once a functional recognition specificity has evolved, it would be advantageous to maintain it in the population, either as an expressed gene or cryptically if there is a cost to expressing numerous R genes.
There are little data on how many R genes are expressed and whether there is a cost to expressing numerous R genes. At least 5 of the 15 copies of M paralogs are expressed (Ellis et al. 1997). Also, at least half of the Dm3 paralogs are expressed (Meyers et al. 1998b; B. Meyers, K. Shen, and R. Michelmore, unpubl.). The large sequence divergence that occurs 5′ to paralogs suggests that they may have different expression levels or patterns. The distribution of 5′ sequences and coding regions forXa21 paralogs suggests the possibility of promoter shuffling, although the expression pattern of each paralog was not studied (Song et al. 1997). It seems unlikely that there is a significant cost to expressing an individual R gene as there are numerous such sequences in the genome and each individual message is usually rare. Most studies on the cost of resistance do not compare plants truly differing in a single resistance gene; as discussed above, all plants express numerous R genes, although the majority cannot be recognized experimentally. However, there may be some cost because expression of NBS–LRR genes from strong promoters is often deleterious (Mindrinos et al. 1994); although this has now been achieved forL6 (Ellis et al. 1997). Also, there is presumably an aggregate cost to expressing many R genes, otherwise such genes would tend to take over the genome.
The Role of Somatic Variation
Somatic variation is a key component in generating the broad and flexible array of specificities exhibited by the vertebrate immune system. In the absence of a circulatory system in plants, somatic variation would, on first consideration, not seem advantageous. However, the plastic and compensating growth form of plants would allow somatic sectors with new resistances to survive and contribute to the overall fitness of the plant. This would be particularly advantageous in clonally propagated species as well as long-lived species such as trees that have generation times far longer than those of potential pathogens. It will be interesting to compare variability in the specificity determining domains of R genes between different parts of long-lived individuals.
Consequences to Breeding for Disease Resistance
There are many useful resistance specificities available. Some may be cryptic within the cultivated species; others are located in wild species. When resistance is introgressed using classical breeding procedures, it is likely that many resistance genes are being introduced simultaneously, replacing those in the recipient genotype. Apparent nonhost/race nonspecific resistance may be, at least in some cases, the result of naturally occurring pyramids of race-specific genes. Multiple resistances to Cladosporium fulvum were detected within the Cf9 haplotype (Parniske et al. 1997).
A current challenge is to dissect these complex families at the molecular level and access the useful variation transgenically. This would allow the transfer of resistance across sexual incompatibility barriers. Also, when we can understand the mechanisms generating new resistance specificities, we can attempt to emulate them ex planta to evolve genes with altered ligand-binding characteristics and providing more effective resistance or new specificities.
Conclusion
Plant resistance genes provide numerous opportunities for studying diversifying selection and the evolution of multigene families. Much of the current theory has been developed from studies on the evolution of the vertebrate immune system. Studies on R genes will provide valuable insights into the evolution of such multigene families. There are numerous R genes in every plant species; it is easy to generate large experimental populations and to select for rare genetic events. Also, the genetic events occurring during the coevolution of plants and pathogens can now be analyzed by comparing clusters of resistance genes using natural populations.
Clusters of R genes are reservoirs of variation for resistance specificities rather than rapidly evolving, dynamic groups of genes. Some genes may be expressed but not functional against current pathogen populations. Other specificities may not be expressed and therefore cryptic but accessible through a variety of genetic events. The majority of R genes are comprised of arrays of hypervariable potential ligand-binding sites. Interallelic recombination within these arrays may be the primary mechanism generating rapid variation in binding specificity. Some unequal crossing-over occurs and is important; however, it is infrequent and not the primary mechanism underlying the rapid evolution of new specificities. The presence of multiple complex clusters of R genes, each with arrays of potential ligand-binding sites (Fig. 2), suggests how plants can generate and maintain large numbers of resistance specificities against ever-changing pathogen populations.
Glossary
As this review draws on ideas from population genetics to structural biology, the terminology used may not be familiar to a general readership. Therefore, at the suggestion of several reviewers, we include these definitions. Further discussion of these and related terms can be found in Li (1997) or Creighton (1993).
The terminology for describing clustered multigene gene families is complex, particularly when structural rearrangements have occurred. For the purposes of this review the terms are used in the following way:Cluster is used to describe a genetically localized group of genes. Gene refers to a single member of a multigene family; each gene occupies a unique position within the cluster.Alleles refer to alternative forms of a single gene and not to paralogs. Homologs are all genes of related sequence and presumably function. Orthologs are genes separated by a speciation event and occupy allelic positions within the cluster.Paralogs are genes that have arisen by duplication events and in this review usually refer to members of a single cluster. Thehaplotype is the aggregate allelic composition across the cluster.
Nonsynonymous nucleotide substitutions result in changes in the amino acid sequence of the protein product. Conversely, synonymous nucleotide substitutions do not result in changes at the amino acid level. Their relative ratio (KA:KS ) is indicative of the selection pressure on the region. If there is no predominant selection on the region, KA:KS will be ∼1. Amino acid changes tend to be deleterious to the conservation of protein structure and function; therefore most sequences are underpurifying selection, which selects against deleterious mutations, and have a KA:KS ratio of <1. Occasionally, there is a selective advantage for amino acid diversity; this has been best characterized for the antigen-binding groove of the MHC protein (Hughes and Nei 1988; Li 1997). Such regions will be underdiversifying selection and have a KA:KSratio >1. The birth-and-death process has been proposed for the evolution of the vertebrate MHC and immunoglobulin clusters that involves the expansion or contraction of the cluster (gene duplication or gene loss) by unequal crossing-over and the evolution of individual genes by diversifying selection (Nei et al. 1997).
Proteins are often modular, with distinct functions performed by different regions (domains) of the molecule (e.g., Kuriyan 1993; Campbell and Downing 1994). In resistance proteins, thereceptor domains that bind to molecules originating from the pathogen directly or indirectly (pathogen-derived ligands) are probably distinct from the effector domains that are responsible for initiation of the signal transduction cascade. By inference from the known crystal structure of porcine ribonuclease inhibitor, R genes are hypothesized to contain a binding surface comprised of arrays of β-strands (Jones and Jones 1997; Kajava 1998); these β-strands have amino acids with residues that face outwards (solvent-exposed residues) and conserved aliphatic amino acids with hydrophobic residues buried in the interior of the protein.
Acknowledgments
We thank Brandon Gaut, Rick Kesseli, Scot Hulbert, Dina St Clair, and Stephan Abel for helpful comments on the manuscript. Our work onDm genes was supported by United States Department of Agriculture National Research Initiative grant 95-37300-1571. The research on Pto was supported by National Science Foundation (NSF) Cooperative Agreement BIR-8920216 to Center for Engineering Plants for Resistance Against Pathogens (CEPRAP) and by CEPRAP corporate associate, Sandoz Seeds. Partial support for B.C.M. was provided by a NSF Graduate Research Fellowship.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL rwmichelmore{at}ucdavis.edu; FAX (530) 752-9659.
- Cold Spring Harbor Laboratory Press








