
Toward a Complete Human Genome Sequence
Abstract
We have begun a joint program as part of a coordinated international effort to determine a complete human genome sequence. Our strategy is to map large-insert bacterial clones and to sequence each clone by a random shotgun approach followed by directed finishing. As of September 1998, we have identified the map positions of bacterial clones covering ∼860 Mb for sequencing and completed >98 Mb (∼3.3%) of the human genome sequence. Our progress and sequencing data can be accessed via the World Wide Web (http://webace.sanger.ac.uk/HGP/ orhttp://genome.wustl.edu/gsc/).
Over the past few years, a revolution in large-scale DNA sequencing has resulted in determination of the complete sequences of a range of viruses, bacteria, and yeast, and the much larger genome (100 Mb) of a metazoan, the nematode wormCaenorhabditis elegans, is on target for completion in 1998. From the genome sequences, near-complete catalogs of genes and gene structures are emerging for these organisms. This in turn opens the way for systematic studies of gene function. Comparison between genome sequences also offers exciting prospects, as functionally important sequences are conserved in evolution and may be identified by their similarity.
The experience gained in these pioneering projects led us to propose in 1995 that a human sequence map of the entire 3000-Mb human genome (comprising precisely mapped sections of sequence determined to at least 99.9% accuracy) could be determined using existing technology (Marshall 1995). The task could be completed by 2005 and, therefore, within the time frame originally proposed for the human genome. Economies of scale, automation of some steps in the sequencing process, and ongoing technical developments would drive costs down and increase throughput sufficiently to enable completion of the task within 10 years. However, international discussion modified this plan, and it was agreed that the full sequence should be determined to 99.99% accuracy and without gaps. The consensus was to proceed on a chromosome-by-chromosome basis, and to ensure optimal coordination of all large and small groups worldwide by positioning all the efforts on a publicly available map.
A number of difficulties were anticipated in this approach. Could the work be carried out on the scale required? Could human genomic sequence be finished to the same standard as that of the nematode? Could the necessary clone resources and maps be constructed? Would costs decrease sufficiently?
As part of the debate, alternative (less costly) approaches to study complex vertebrate genomes were proposed by others. These included large-scale sequencing of expressed sequence tags (ESTs) (Adams et al. 1991; Hillier et al. 1996) to define comprehensive sets of genes and complete sequencing of the smaller (400-Mb) genome of a model vertebrate, the puffer fish Fugu rubripes (Brenner et al. 1993). However, such studies cannot provide a complete picture of the human genome. The data from these projects will be much more valuable when aligned to the human genomic sequence to find and annotate genes (Ansari-Lari et al. 1998).
In contrast, the complete human genome sequence will provide the ultimate map of the genome. Every piece of genetic information (genes, promoters, enhancers, CpG islands, genetic markers, etc.) will be placed on a single metric to an accuracy of 1 base, provided by the contiguous sequence. The sequence will contain the information necessary to define all the genes and all other biologically important sequences in the genome.
Over the past 3 years, we have developed a strategy to determine the sequence of a substantial portion of the human genome. A large-scale program has been established, and together, our two laboratories have finished 98 Mb of human genomic sequence (∼3.3% of the genome). Based on our progress so far, we are confident that the prospects are excellent for sequencing the rest of the human genome by 2005, as part of an international collaborative effort involving many laboratories (see Table 1). In this article, we describe our work to date and discuss the problems we have solved so far and the issues that still must be addressed.
International Consortium of Laboratories Involved in Large-Scale Sequencing
Determining the Human Genome Sequence
A key to the success of the C. elegans sequencing project was the availability of an almost complete physical map of the genome in overlapping cosmids that were joined by bridging yeast artificial chromosome (YAC) clones (Coulson et al. 1986, 1988). A minimally overlapping subset of cosmids was selected from the available contigs for most of the sequencing, and this is now being complemented by shotgun sequencing of selected YACs to finish the project.
In contrast, the most comprehensive physical maps of the human genome have comprised sets of overlapping YACs ordered on the basis of shared landmark content [chiefly sequence-tagged sites (STSs) (Olson et al. 1989)]. The contigs were positioned using markers that were also present in the available genetic (Murray et al. 1994; Dib et al. 1996) or radiation hybrid (RH) maps of the human genome. The average STS spacing of these combined maps is ∼1/250 kb for the whole genome maps (Chumakov et al. 1995; Hudson et al. 1995), and up to 1/70 kb for selected single chromosomes (Chumakov et al. 1992; Foote et al. 1992;Collins et al. 1995; Doggett et al. 1995; Gemmill et al. 1995; Krauter et al. 1995; Bouffard et al. 1997; Nagaraja et al. 1997). Although long-range continuity was obtained with these maps, human YACs were not considered suitable substrates for large-scale genomic sequencing in general, because of the high degree of chimerism and the instability observed in a large proportion of the YAC clones (Green et al. 1991;Nagaraja et al. 1994). Therefore, these early maps must be converted to a practicable reagent to support the large-scale sequencing of the human genome. More recently, RH mapping has been used to construct a physical map of the human genome, containing 30,181 unique gene-based markers (Deloukas et al. 1998). Mapped markers from both the gene map and the YAC based maps form the basis for the construction of bacterial clone maps for sequencing.
For the human genome, an important breakthrough for building sequence-ready maps was the development of the new large insert (up to at least 200 kb) bacterial- and P1-artificial chromosome (BAC and PAC, respectively) (Shizuya et al. 1992; Ioannou et al. 1994) cloning systems and the construction of comprehensive human genomic libraries. There are currently a number of libraries available, with a combined 60-fold representation (see Table 2), and more are presently under construction (P. de Jong and U.-J. Kim, pers. comm.). Clones from these libraries are stable and contain few rearrangements on the basis of the data available so far (Shizuya et al. 1992; Ioannou et al. 1994).
Bacterial Clone Libraries That Are Currently Available for Human Sequencing
Our strategy for constructing sequence-ready maps (Fig.1) consists of screening for bacterial clones using a high density of STSs (15/Mb on average). These markers were initially taken from the available YAC maps (for chromosomes 2, 7, 22, and X) and were supplemented with all other markers in the public domain. For chromosomes 1, 6, and 20, markers derived from ESTs (Hillier et al. 1996) and flow-sorted chromosomes (Ross and Langford 1997) were used together with existing markers to assemble integrated maps by RH mapping (Cox et al. 1990; Walter et al. 1994; Mungall et al. 1996) (maps are available at http://www.sanger.ac.uk/HGP/Rhmap). The bacterial clones are assembled into contigs by comparative restriction fingerprint analysis (Coulson et al. 1986; Olson et al. 1986; Gregory et al. 1997; Marra et al. 1997) and landmark content mapping (Green and Olson 1990). The nonuniform distribution of markers means that many gaps remain at this stage, but contigs are then extended and joined by the generation of new markers at their ends. Some larger gaps are also filled using region-specific probes generated from bridging YAC clones. A minimally overlapping subset of the bacterial clones is selected for sequencing by manual inspection of the overlapping fingerprints relating to each clone. All selected clones are verified by ensuring that all available restriction patterns, landmark content, and fluorescent in situ hybridization data for each clone are entirely consistent. This general approach has two advantages over other methods such as whole-genome shotgun (Weber and Myers 1997; Venter et al. 1998) or end-sequencing BAC clones followed by walking (Venter et al. 1996). Neither of the latter approaches can be coordinated efficiently to minimize overlap between collaborating groups, nor do they make use of all the available map information that helps verify order and clone fidelity (see also Green 1997). Nevertheless, the data produced from such efforts, if released into the public domain, will be synergistic with clone-based approaches (Waterson and Sulston 1998).
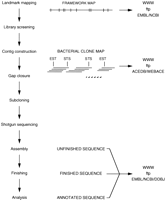
Strategy for determining the human genome sequence. Sequence-ready maps are constructed by screening for bacterial clones using a high density of STSs (15/Mb on average). Bacterial clones are assembled into contigs by comparative restriction fingerprint analysis and landmark content mapping. Contigs are extended and joined by generating new markers at their ends or using region-specific probes generated from bridging YAC clones. A minimally overlapping subset of clones is selected for sequencing, after ensuring that for each clone all available restriction patterns, landmark content, and fluorescent in situ hybridization data are consistent. DNA fragments are derived from the bacterial clones and subcloned into bacteriophage M13 or plasmid vectors. The aim is to achieve an average of approximately six- to sevenfold coverage in high-quality bases for each base of the bacterial clone insert from 2500 reads. More reads may be added to facilitate finishing of difficult clones and to compensate for the occasional higher than expected rates of failure (sequencing and gel failures, vector reads, and contaminating sequences). These can generally be assembled into 2–10 contigs representing >98% of the bacterial clone. This preliminary consensus, or unfinished, sequence is subjected to automatic and manual editing, and additional directed sequence reads are performed to close the remaining gaps and to resolve all ambiguities, thus providing the finished sequence. The entire sequence is checked and analyzed using a variety of computer tools and manually annotated. At all stages, raw data, analysis, and status information are publicly available via the internet. Full experimental details can be found via URL http://www.sanger.ac.uk/HGP/methods/ andhttp://www.genome.wustl.edu/ (see also Gregory et al. 1997; Leversha 1997; Dunham et al. 1998).
Our joint mapping progress to date is summarized in Table3. The first phase is almost complete: Where required, landmark maps have been completed for almost all of the 1100 Mb currently in hand (on eight chromosomes) to a density of 15 markers per megabase. The second phase, BAC and PAC clone isolation, is also proceeding rapidly, and coverage of ∼860 Mb has been obtained as of September 1998. Furthermore, no marker tested against all libraries has failed to find a positive clone. The third phase, walking and gap closure, is now in progress in several regions. For example on chromosome 22q (Fig. 3c, below), seven contigs now contain ∼20.2 Mb, and all the remaining ends are still actively being extended. The longest contig is estimated to be ∼14.7 Mb. Taken together, our experience confirms that sequence-ready maps of long contiguous stretches of the human genome can be provided using current resources.
Joint Mapping Progress
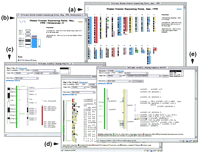
The plans and commitments of each sequencing laboratory (Table 1) are shown as color bars adjacent to schematic images of each chromosome (a). The information is taken from the HGSI athttp://www.ncbi.nlm.nih.gov/HUGO/, except for chromosomes 21 and X, where the display reflects the information of the respective consortia (chromosome 21, http://www-eri.uchsc.edu/chr21/consortium.html and the X chromosome, http://www.sanger.ac.uk/HGP/ChrX/xchrom1.shtml). We are aware that there are additional plans not yet registered on the HGSI, which are not illustrated here. Clicking on the schematic ideogram displays an enlarged view of just that chromosome (b). Clicking on the left most colored bars follows links to the corresponding laboratory Web sites. Clicking on the adjacent and narrower bars follows links to map views, such as the status of the chromosome 22 map (c). From here, access can be gained to displays of the bacterial clone contigs, e.g., contig 22 in dshowing the sequence status of each clone; and from there, to displays of the sequence annotation, e.g., clone dJ434P1 in e. Images shown in c–e are live views provided via webace2, a WWW front end to ACeDB.
Our sequencing strategy is essentially the same as that which has proved so successful in the C. elegans project and proceeds in two phases. In an initial shotgun phase, DNA fragments (1.4–2.2 kb) derived from a bacterial clone are subcloned into bacteriophage M13 or plasmid vectors (Bankier et al. 1987) for sequencing by the chain terminator method (Sanger et al. 1977) using both types of fluorescent chemistry (dye-labeled primers and dye-labeled terminators) (Prober et al. 1987; Smith et al. 1987; Lee et al. 1992). Sequence reads are assembled into typically 2–10 contigs representing some 95% of the bacterial clone, and the assembled sequence is referred to as “unfinished.” In a subsequent directed phase termed “finishing,” the preliminary consensus sequence is subjected to automatic and manual editing, and additional directed sequence reads are performed to close the remaining gaps and to resolve all ambiguities. The aim is to determine all bases using reads on both strands or by different chemistries on at least two subclones. The rare exceptions are reviewed and noted. The entire sequence is also checked using a variety of software tools, which flag potential editing errors and ambiguities. Finally, an in silico restriction digest of the finished sequence is compared with the experimental digest of each clone.
To date, we have finished 98.4 Mb of human sequence. This constitutes ∼3.3% of the genome and 50% of the world output so far (190 Mb in segments >10 kb; see Fig. 2). All human genomic sequence is being finished to a defined quality of 99.99% accuracy, on a clone by clone basis, leaving no gaps in the sequence of each clone. The accuracy is achieved partly because of the redundancy of raw data in the multiple reads obtained by the shotgun approach and partly as a result of the well-developed finishing process, which includes visual checking of every sequence with a highly trained eye. At this early stage, many sequences are present as isolated clones; contiguity will increase as walking continues.
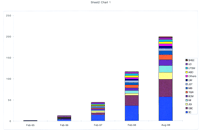
Growth in human sequence output (>10-kb segments only) in the past 3 years. Data are taken from Genome-MOT (http://www.ebi.ac.uk/∼sterk/genome-MOT/) (Beck and Sterk 1998), from the NCBI, and from annual reports of the International Strategy Meetings on Human Genomic Sequencing. (See Table 1 for guide to abbreviations of each laboratory name.)
Sequencing Methods and Technology
Throughout the project it is essential to be able to generate high quality data consistently and on a large scale. The system must be both robust and flexible enough to accommodate changes in technology or staff while continually improving overall performance. The process is organized in a modular fashion, and automation is integrated where it yields significant improvements (in either throughput or accuracy) over manual procedures. This blend provides important advantages over complete automation. The staff undertakes complex decision making, introduces and monitors appropriate new technology, provides feedback, and acts as a source of continual training, growth, and improvement at all stages of the process. The continued introduction of improvements will allow the existing methods to be scaled up to provide the necessary sequencing capacity to meet our targets.
Over the past 2 years, major increases in efficiency of sequence output have been brought about by improvements in three main areas: automation of individual processes, improvement of the underlying biological and chemical systems, and software development.
One continuing factor in increasing throughput is the number of useful bases obtained per sequencing instrument per run. Changes in DNA sequencing chemistry have led to substantial improvement in the quality and read length of the primary sequence, with no extra labor investment per sample. These changes include the introduction of fluorescent energy transfer dyes (Smith et al. 1985; Ju et al. 1995; Rosenblum et al. 1997) for more sensitive labeling of DNA and engineered thermostable polymerases (“FY”) that are more processive and less discriminant in incorporating dideoxy nucleotides (Tabor and Richardson 1995). A complementary approach has been to increase the number of samples loaded per run. Hardware (ABI 377 automated sequencers, Perkin-Elmer) and software have been modified to increase the number of lanes from 48 to 96 lanes. This increase in capacity requires no increase in gel preparation or sample manipulation, thus reducing the labor per sample. Further improvements, such as capillary electrophoresis systems (Huang et al. 1992), will reduce effort for this step more substantially.
Other important steps that have been automated include the picking of clones for sequencing templates (3000 are required per 100-kb PAC or BAC clone) and selection of templates for configuration of custom finishing reactions. Ninety-six-channel aliquotting devices are in use routinely for the large-scale setup of shotgun sequencing reactions and for template preparation. Further automation of these and other processes is in progress and promises to reduce both labor and error rate further.
The most difficult problems encountered in finishing human genomic sequence are regions that are highly repetitive or GC rich, and it has been necessary to develop new approaches to solve them. For example, GC-rich regions may possess secondary structures that significantly affect the processivity of the DNA polymerase used. The result is an area of low-quality data from which the exact sequence of the region cannot be accurately deduced. Some of these regions can be resolved simply by using various dye-labeled terminator chemistries. Other, more difficult GC-rich regions have been overcome by isolating a restriction fragment or PCR product spanning the region, shearing it into small fragments by sonication, and using these to construct a small-insert library of very short random subclones (e.g., 100- to 200-bp inserts) for sequencing (McMurray et al. 1998). A relatively small sampling of the small-insert library (typically <100 subclones) provides adequate coverage of the region, and the small insert size usually ensures the disruption of the associated secondary structure that previously prevented the generation of high-quality sequence data.
Software developments have played a key role in increasing throughput in both the shotgun and finishing phases. A major bottleneck in analysis and transfer of the data generated by the DNA sequencers has been resolved by the development of UNIX-based software for lane tracking and gel image processing [GELMINDER (unpubl.)] and base calling [PHRED (Ewing et al. 1998; Ewing and Green 1998)]. Contaminating bacterial or yeast sequences (from host cell DNA carried over during subcloning), vector sequences, and low-quality data are all filtered out automatically, and a statistical summary to monitor data quality is calculated [ASP (unpubl.)]. The remaining sequences are then assembled automatically [PHRAP (P. Green, pers. comm.)]. Another important bottleneck has been the manual review of assembled, unfinished sequence, which is required to select the additional sequencing reactions for gap closure and problem solving. Much of this decision-making process is now done automatically by the program FINISH (Dear et al. 1998), which provides lists of subclone names and types of sequencing reaction required, in conjunction with powerful sequence contig editors (Bonfield et al. 1995; Gordon et al. 1998).
Analysis, Annotation, and Data Release
All finished and unfinished assembled sequence is made available immediately on the internet, via local FTP sites and at the public sequence databases [the EMBL library at the European Bioinformatics Institute (EBI) and the National Centre for Biotechnology Information (NCBI)]. All maps and sequence annotation are available on the World Wide Web (WWW) as graphical displays in the database ACeDB (Durbin and Thierry-Mieg 1991). To provide easy access to the status of the international program, we have mounted a graphical view of the human chromosomes (see URL http://webace.sanger.ac.uk/HGP/ orhttp://genome.wustl.edu/gsc/) that serves as a common entry point to all subsequent levels of information at all the web sites of the different participating laboratories (Fig. 3a). These pages are available by FTP and can be freely mirrored at any WWW site. The international status is provided by the Human Genome Sequence Index (HGSI) displayed at the NCBI web site (Bentley et al. 1998) (http://www.ncbi.nlm.nih.gov/HUGO).
In bacterial and yeast genome sequences, computational analysis and annotation of genes have led to near complete catalogs of known or potential genes. Gene structures are readily identified, as the base composition of coding and noncoding DNA sequence is easily distinguished and transcriptional start points match well-conserved motifs. There are few, if any, introns and a much lower proportion of noncoding sequence in these genomes than those of more complex organisms. In contrast, when applied to human sequence, predictions by these ab initio methods are much less reliable, and so our analysis relies much more heavily on the results of homology searches against all available DNA and protein sequences.
Annotation of the sequence is carried out in implementations of the database ACeDB, into which are loaded all the results of gene predictions and sequence searches. A subset of features are annotated and exported to create entries for submission to EMBL or GenBank. ACeDB database files containing all the analysis information divided by chromosome are made available via FTP. A WWW interface to ACeDB (webace) provides direct access to the annotation, using graphical views of all analysis results for any region of the sequences (Fig. 3e).
So far we have identified 673 confirmed genes from analysis of finished, annotated sequence produced in the two centers. This is necessarily an underestimate owing to the limitations of ab initio and sequence search methods and the incomplete sequence databases being searched. Full lists of genes predicted so far can be accessed viahttp://webace.sanger.ac.uk/humace/genes/,http://genome.wustl.edu/gsc/CDS/cds.html, andhttp://genome.wustl.edu/gsc/GENSCAN/genscan.html.
Future Prospects
Through our joint efforts, over 98 Mb of human genomic sequence has been completed and a further 80 Mb is available as assembled unfinished sequence. When combined with the data from other participants in the international effort, the total finished sequence available is 190 Mb as of September 1998. The contribution of all groups is additive (see Fig. 2). Wasteful duplication has been minimized by good coordination of all ongoing efforts, organized relative to a consensus framework map and displayed in the HGSI (Bentley et al. 1998).
Sixty megabases of the finished total was produced by the two centers in the 12 months to September 1998. With additional resources becoming available and further technical improvements, we expect our finished sequence output to rise to 150–200 Mb per year. Additional sequencing capacity will provide a further acceleration in the production of unfinished, assembled sequence. This provides the community with early access to a fast-growing source of valuable mapped genomic sequence information. Production of such a “working draft” of the human genome as unfinished, assembled sequence is projected to be 90% complete by the end of 2001 (Marshall 1998; Wadman 1998).
In common with other centers undertaking large-scale sequencing, we have found that our costs have not fallen as rapidly as we predicted (Marshall 1995). It has not been practicable to address the demands of scaling up the production of high-quality sequence and simultaneously to maximize our cost efficiency. We remain optimistic, however, that over the next 5 years the cost of finished sequence will fall to <15 pence (25 cents) per base.
This work illustrates the speed at which all of the remaining chromosomes can be mapped. The new bacterial clone libraries appear to be highly representative; in some instances they cover regions not represented in available YAC maps, though in rare cases YACs may still provide a valuable complement to them. Walking in a 25-Mb region of chromosome 22 over the past year has reduced the number of contigs from 65 to 7 so far, and progress is active at all remaining ends. This gives the first strong indication that long-range continuity can be obtained in bacterial clones. Therefore we believe that 90%–95% of the human genome sequence can be determined in multimegabase sections of contiguous sequence of high accuracy. The remainder, comprising centromeric and telomeric sequences, satellites, etc., presents special problems and must be treated separately.
It is reasonable to expect that knowledge of the sequence will in itself stimulate progress in computational analysis of the genome. Research will be facilitated by a rapidly increasing data set for testing, leading to improved algorithms. Sequence comparisons with other genomes and within the human genome itself will be revealing. The sequence will provide the tools for biochemical investigations that will check the computer predictions and provide information for further development.
Despite the small fraction of the human genome (6.3% worldwide, 3.3% from the two centers) completed so far, the rapid release of finished and unfinished sequence data is already aiding important new discoveries. Over the last 6 months, sequence has been downloaded >80,000 times from the Sanger Centre or Genome Sequencing Center FTP sites, and our BLAST servers to search human sequence were accessed from over 2000 different computers in external laboratories worldwide. Many new genes involved in human diseases have already been found and characterized much faster using the available genomic map and sequence data. Notable examples include genes involved in breast cancer (Wooster et al. 1995), X-linked retinoschisis (Sauer et al. 1997), Pendred syndrome (Everett et al. 1997), and X-linked lymphoproliferative disease (Coffey et al. 1998; Sayos et al. 1998).
Important as they are, however, these short-term rewards are small in comparison with the long-term benefits, in terms both of biological understanding and of medical advances. Knowledge of the human genomic sequence in its entirety will provide a firm foundation for all future biomedical research. Yet, it can be collected for an investment of 0.01% of the cost of health care over the next 7 years. The international coordination and the agreement to free and open data release, which have been achieved through the Bermuda agreement (Table1), provide the means to reach this goal.
Acknowledgments
This paper describes work carried out by staff at the Sanger Centre, the Genome Sequencing Center, and collaborators. Readers are asked to refer to the appropriate WWW site for experimental procedures and further details. We gratefully acknowledge the support of The Wellcome Trust (grant 048880), the Medical Research Council of Great Britain and Northern Ireland, and the National Institutes of Health. We are grateful to all those who have assisted in the preparation of the manuscript and its figures.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 E-MAIL ; FAX 44-1223-496919.
-
- Received July 7, 1998.
- Accepted October 6, 1998.
- Cold Spring Harbor Laboratory Press








