
More BLAST for the Buck
Time and money—they are the essential, yet most often, the limiting factors in people’s lives. Sequence comparisons via computer searches are certainly no different. For the sequence similarity search, time is determined by how long it takes to compare your sequence of interest to the burgeoning sequence information available, whereas matches found are the currency. Of course, not just any sequence match is worthwhile—only matches that are likely to have biological significance; your money should be worth something. To obtain such matches requires a program that is both sensitive (able to pick up relevant matches of weak similarity) and selective (provide a list of matches that are all biologically relevant). The balance between these two can greatly increase the time it takes to run a program—too high a sensitivity and it will take too much time and produce too many nonbiologically relevant sequences; too low a sensitivity and the program will run much faster, but meaningful matches may be lost. The BLAST program has been the primary method for doing database searches to detect related sequences for the purpose of classifying proteins into functional families and identifying unknown functional units. Now, a paper by Altschul et al. (1997) provides modifications to the current algorithms in the BLAST program that save the user time and provide far greater return on the investment.
The new algorithms provide three important changes to the current BLAST program. The first is that the program can run approximately two to three times faster—important in a world of rapidly expanding sequence information. The second is that the program can now allow the production of gapped alignments, which often result in detection of biologically relevant matches that were originally overlooked. The third is the development of a motif or profile searching program within the BLAST system. Motif searches almost always provide the most sensitive analyses.
Sequence search programs have been around for quite a while and are an essential tool for any scientist dealing with protein or DNA sequences of unknown function. The basic strategy behind search programs is to compare one sequence of interest with another sequence(s) of interest and assess the amount of similarity between the two. Over a particular length of sequence, matches provide a positive score, whereas mismatches are penalized relative to an exact match. The basic scoring strategy is developed taking into account how conservative such a change is with regard to protein or DNA changes through evolution. During a comparison, a total score is tallied: the higher the score the greater the similarity of the compared sequences. High scoring sequences are presumed to have biological relevance to the sequence of interest, although there are some caveats to this, such as the presence of repeat elements of no particular biological importance. Additionally, areas of sequence homogeneity (e.g., AT- or GC-rich regions) can also produce a high score that carries no biological relevance.
BLAST to Begin With
The original BLAST program (Altschul et al. 1990) essentially worked as follows: The algorithm compared the sequence of interest to another sequence or database by comparing a “word” (a short, specified length of sequence) within the sequence of interest to “words” in other sequence(s). Each time the word (w) matched another with a score equal to that selected by the user (T), the program extended its analysis to the sequence surrounding w. The extended analysis generated a score indicating how similar this region was overall. The extension continued until the score began to fall below a particular level. The final score provided for a region was the best score that could be obtained without further extension or trimming. This score was recorded, and the search continued to other areas and/or sequences. The value T sets the level of sensitivity. The lower the value for T, the more matches there will be—but the analysis then takes longer as more regions must be analyzed for extended similarity. Careful assessment ofT is required to maintain high selectivity without too great a loss in sensitivity.
The original BLAST program was a breakthrough in database analyses as it enabled far faster searches than previous programs, such as FASTP or FASTA (Lipman and Pearson 1985; Pearson and Lipman 1988) while still not losing sensitivity.
Twice the Speed—All the Sensitivity
Gaining twice the speed and increased sensitivity at a time when databases continue to loom ever larger as more and more sequence data pour out of sequencing centers comes about through a surprisingly simple mechanism (Altschul et al. 1997). Essentially, extension of sequence similarity is only triggered in situations where two matching words are detected, rather than one. The reasoning here is that sequences that will hold a high similarity score (and be biologically relevant) are likely to have at least two nonoverlapping short regions of sequence (w) that will give a score of at least T. Thus, the algorithm still specifies that a search is made for w with a score equal to T. However, unlike the original BLAST, upon identification of the firstw ⩾ T, no extension is made; instead, the analysis continues, searching for another w within a certain distance of, but not overlapping, the first. Upon identification of a second w ⩾ T, an extension examining the surrounding sequence occurs. In the two-hit method, the value ofT is set lower to make up for the loss of sensitivity attributable to the requirement for two matching words. This increased sensitivity results in the identification of 3.2 times as many significant w in the two-hit method, but only ∼0.14 hit extensions are generated given the lower number of matched words that occur within appropriate range of another match. As it is the extension that takes up the bulk of time in sequence comparison (>90% of the time), the two-hit method, without any loss of sensitivity, is by far faster.
Filling in the Gaps
Identifying gapped alignments was theoretically possible in the original BLAST program (Altschul et al. 1990); however, only at much greater analysis time, and the importance of the matches remained questionable. Furthermore, the algorithm for the original BLAST restricted the gapped search alignments to a specific region (Chao et al. 1992) surrounding the region of similarity. This constraint meant that a more optimal gapped alignment that might be present beyond the confines of this boundary would be missed.
The gapped alignment algorithm provided by Altschul et al. (1997)provides a much more dynamic approach to the problem. Here, this constraint on the path to examine is removed. The primary
restriction set is that only those regions where local alignment does not fall a certain amount below the best score yet seen
can be considered for a gapped extension. The gapped alignment proceeds outward in any direction from the two centermost residues
of the highest scoring 11-residue region within the area. The investigators concede that more advanced methods for selecting
the seed are possible; however, test runs suggest that this method is quite effective. The more troublesome elements of the
gapped alignments are two. First and easily addressed is that the gapped BLAST extension time can take ∼500 times that for
an ungapped extension. The investigators correct this by simply raisingT so that gapped extensions occur only
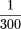 of the time that an ungapped extension occurs. The second difficulty is that the statistical parameters of the program cannot
be computed during the run. Thus, the program is limited to scoring systems for which estimates of these parameters, λg and Κg, are already available. Use of simulated parameters are reasonable; however, this places a constraint on the program in terms
of the scoring system that can be used. A simulation of λg and Κgmust be provided for alternative scoring systems.
of the time that an ungapped extension occurs. The second difficulty is that the statistical parameters of the program cannot
be computed during the run. Thus, the program is limited to scoring systems for which estimates of these parameters, λg and Κg, are already available. Use of simulated parameters are reasonable; however, this places a constraint on the program in terms
of the scoring system that can be used. A simulation of λg and Κgmust be provided for alternative scoring systems.
Motifs Made Easy
Motif searches always provide the most sensitive as well as selective searches, as they can detect very weak, yet important regions of similarity. The difficulty, however, is that creating the consensus profile to be identified can be extremely time consuming and often requires greater understanding of the search program by the user than is optimal for satisfactory general use. The final additional modification that Altschul et al. (1997) made to the program provides the user the ability to automatically create profiles to be used in a BLAST search. PSI–BLAST (forposition-specific iterated BLAST) essentially is an add-on to the beginning of the BLAST program that creates a position-specific matrix score automatically and the matrix is compared with sequences within the database. PSI–BLAST run time is slightly increased from that of the new BLAST program but still remains faster than the original, and the sensitivity of the searches is superior. Particularly useful is that running the PSI–BLAST program multiple times can further refine the profile and increase the sensitivity of the program. Several other motif search programs are also now becoming available. Henikoff and Henikoff (1997) present a program that, instead of modifying the BLAST program itself, runs BLAST by comparing an artificially derived single sequence that effectively takes the place of the position-specific matrix generated in PSI–BLAST.
Additionally, on the Saccharomyces cerevisiae genome database (SGD) (http://genome-www.stanford.edu), a motif search program called Patmatch is also under development but is currently available for use. The program allows one to search the entire yeast protein and DNA database for specific motifs of interest. It is set up to be quite user friendly (http://genome-www.stanford.edu/Sacch3D/patmatch.html). The current format of the motif searching program provided by Altschul et al. (1997), like the program available at SGD, will likely change over time. As the investigators indicate, additional optimization could be applied. However, as evidenced from the results of the examples tested, PSI–BLAST as well as the two other BLAST modifications will clearly will be of great use to the community for its ease of use, speed, and enhanced sensitivity.








