
The Complex Mutation Pattern of a Microsatellite
Abstract
DQCAR is a (CA)n microsatellite located in the HLA class II region and tightly linked toHLA–DQB1. Previous studies showed a strikingly low level of size variation in DQCAR alleles within an extensive subfamily of HLA–DQ subtypes (DQ1). DQCAR alleles in non-DQ1 subtypes showed a higher degree of size polymorphism. In this study sequence analysis demonstrates that DQ1- associated DQCAR alleles have a single C → A nucleotide substitution interupting the CA repeat array. Frequent CA → GA mutations are also observed in DQ1-associated microsatellites with identical allele sizes. In contrast, DQCAR alleles associated with non-DQ1 haplotypes display a perfect CA repeat sequence and the variation in allele size is attributable only to differences in the number of CA repeats. Our results imply that several mutational mechanisms are involved in the generation of allelic diversity within the same microsatellite locus. The possibility of different mutation rates in the same locus should to be taken into account when using these markers in evolutionary and disease studies.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos.U96944–U96962 and S87165.]
Microsatellites are abundant in mammalian genomes (Weber 1990;Stallings et al. 1991) and have attracted an enormous amount of interest in recent years. Instability of trinucleotide repeats is involved with certain neurodegenerative conditions (for review, see Rosenberg 1996), suggesting that underlying mutational mechanisms may be responsible for genetic morbidity. Polymorphic dinucleotide repeats have been used extensively for gene mapping (Dib et al. 1996), forensic identification (Edwards et al. 1992) and studying the history of human population (Bowcock et al. 1994; Deka et al. 1995).
In spite of their popularity as a genetic tool, the mechanisms underlying microsatellite allelic diversity are still poorly understood. Estimations of mutation rate for microsatellites range between 10−2 and 10−5 per generation (Edwards et al. 1992; Mahtani and Willard 1993), with Weber and Wong (1993)observing that dinucleotide repeats have a lower average mutation rate than tetranucleotides (5.6 × 10−4 and 2.1 × 10−3, respectively). Most of the observed changes in microsatellite allele sizes are attributable to deletion or addition of a few repetitive units (Straub et al. 1993; Deka et al. 1995). In principle, this pattern of mutation fits with a simple stepwise mutation model (Ohta and Kimura 1973) but might not account for all the variation observed (Shriver et al. 1993; Valdes et al. 1993; Deka et al. 1995). A two-phase model that allows for bigger differences in repetitive units has also been proposed (Di Rienzo et al. 1994). Strand slippage during replication has been suggested as the most likely mechanism involved in the generation of microsatellite mutations (Levinson and Gutman 1987; Schlötterer and Tautz 1992;Strand et al. 1993).
Most of the studies involving microsatellites have analyzed a large number of loci. Detailed sequence analyses of mutation patterns at individual loci have been performed especially for trinucleotides associated with diseases (Rosenberg 1996) and some tetranucleotides (Mahtani and Willard 1993; Talbot et al. 1995). Minisatellites have also been analyzed (Jeffreys et al. 1994). Dinucleotides, however, have received less attention.
Previously, we studied a (CA)n microsatellite (DQCAR) located in the human leukocyte antigen (HLA) class II region, ∼12 kb centromeric to HLA–DQA1 and 1–2 kb telomeric to the HLA–DQB1 gene (Satyanarayana and Strominger 1992). DQA1 and DQB1 encode for the respective α and β chains, which form the heterodimeric protein HLA–DQexpressed in the surface of cells involved in the immune response. Fourteen different DQCAR alleles were identified and these were found to be in tight linkage disequilibrium with HLA–DQ(Macaubas et al. 1995; Mignot et al. 1995; Jin et al. 1996). Extremely low size polymorphism was found within DQCAR alleles associated with DQ1, the predominant HLA–DQ subtype in several populations (Imanishi et al. 1992). The DQCARrepeat associated with the other DQ subtypes (DQ2, DQ3, andDQ4) showed both larger numbers and sizes of alleles (Macaubas et al. 1995). In this study we carried out a sequence analysis ofDQCAR alleles to investigate the basis for the observed allelic differences between DQ1 and non-DQ1haplotypes.
RESULTS
The phylogeny of DQB1 alleles is shown in Figure 1(left). There are two groups of alleles—theDQ1 subgroup (DQB1*05 and DQB1*06) and non-DQ1 (DQB1*02, DQB1*03, and DBQ1*04). The typing of 2140 chromosomes (Fig. 1, column 2), showed that in the non-DQ1 subfamily, more DQCAR alleles per DQB1 subtypes were found (Fig. 1, column 3) in comparison with theDQ1-associated subgroup. Non-DQ1-associatedDQCAR alleles also have longer microsatellite sizes (Macaubas et al. 1995; Jin et al. 1996). The exceptions were DQCARalleles associated with DQB1*0201, with only two short microsatellite alleles detected in >150 chromosomes typed.
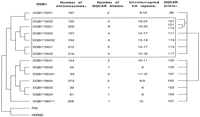
Phylogenetic relationships of DQB1 alleles (left) andDQCAR alleles (right) based on their sequence data. Phylogenies of 14 DQB1 and 15 DQCAR alleles were reconstructed as detailed in Methods. (a) Number of chromosomes typed for DQB1 and DQCAR in a mixed population. (b) Number of DQCAR alleles associated with each DQB1 allele in the mixed population. (c) Number of uninterrupted CA repeats for each DQCAR allele observed in the mixed population, deduced from the sequenced alleles on Table 1. (d) The DQCAR alleles represented in the tree are the sequenced from Table 1. Areas with high DQCARsize variation (more than two DQCAR alleles found in association with a lineage) are boxed.
At least one DQCAR allele from 15 DQB1 lineages was then subcloned and sequenced. All DQCAR alleles associated with the DQ1 subfamily showed a C → A nucleotide substitution in the fifteenth base pair, resulting in a CAAA motif interrupting the CA repeat array (Table 1; Fig.2). In contrast, the microsatellite allele sequences associated with non-DQ1 subfamilies showed an uninterrupted CA repeat motif (Table 1; Fig. 2).
DQCAR Sequences Observed in Association with VariousHLA–DQB1 Alleles
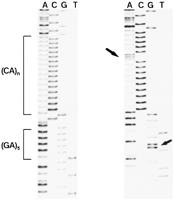
Sequence from two representative cell lines. (Left) Cell line 9072 (DQ4, DQB1*0402, DQCAR117); (right) cell line 9062 (DQ1, DQB1*0603, DQCAR103). The left arrow points to the A nucleotide interrupting the CA repeat sequence in the DQ1 cell line. Note the tetramer GCAA between the (CA)n region and the (GA)5 in the DQ4 cell line. The right arrow points to the G substitution in the flanking region, characteristic ofDQCAR103 and DQCAR107. Sequence of opposite strand confirmed the results in both cases (not shown). Sequence was performed as detailed in Methods.
Non-DQ1-associated alleles differed in size as a result of varying numbers of CA repeats. DQCAR 99 (the shortest allele sequenced) has a repeat array of 9 uninterrupted CA units, andDQCAR 121 (the longest) has a stretch of 22 CA units. Some changes from C → G into the repeat array were also observed in some alleles on both sides of the CA repeat (Table 1).
Each DQCAR allele is composed of a CA repeat, followed by a GA motif. Non-DQ1-associated alleles always show an intervening tetramer GCAA between CA and GA motifs. In DQ1-associated alelles the GA repeat structure lies immediately next to the CA repeat array (Table 1; Fig. 2). The GA repeat motif in non-DQ1-associated alleles is monomorphic, with five repeats of GA (Table 1; Fig. 2). The number of GA repeats varies forDQ1-associated alleles, even when the sizes of two alleles, are identical. For example, DQCAR 103 alleles associated withDQB1*0602 and with DQB1*0603 have the motif (CA)8(GA)4 and (CA)9(GA)3, respectively. The total number of repetitive units for those alleles remains constant (i.e., 12 units) while their composition varies. Using routine genotyping methods, this variation is missed.
The sequence flanking the 3′ end of DQCAR showed subtype-specific variations. DQ1-associated alleles showed a G nucleotide in the fifth nucleotide after the GA run (Table 1; Fig. 2). Non-DQ1 alleles have an A in this position. Sequences fromDQB1*0202 and DQB1*0301 reveal a T in the ninth nucleotide after the GA stretch. All of the other sequences (includingDQ1 sequences) have a G in this position (Table 1).
A phylogenetic tree of DQCAR alleles, based on their flanking sequence, was then constructed (Fig. 1, right). As for DQB1alleles, two groups were observed: DQCAR alleles associated with DQ1 subtypes formed one group, whereas microsatellite alleles observed with non-DQ1 subtypes clustered in a separate group. The number of uninterrupted CA repeats for each DQCARallele observed in the typed population was deduced from the sequence data (Fig. 1, column 4). The non-DQ1-associated DQCARalleles showed a higher number of uninterrupted CA repeats (13–24 CA repeats), compared to DQCAR associated with DQ1haplotypes (between 8 and 12 CA repeats). The only exceptions wereDQCAR alleles associated with DQB1*0201, with 9 and 10 repeats.
DISCUSSION
In the DQCAR locus, three forms of mutations are responsible for the generation of new alleles: (1) simple gain or loss of CA units; (2) a single C → A substitution in the CA repeat array; and (3) GA → CA or CA → GA mutations in the region immediately following the CA repeat array. Only the first mutation mechanism resulted in a change of allele size.
We observed that for DQCAR alleles with perfect CA-repeat runs, the change in allele size is attributable to small differences in the number of repetitive units, mostly by only 2 bp. This pattern of mutation seems to fit with a model of small changes in repeat unit number via a strand-slippage mechanism (Strand et al. 1993). In addition, the increase was more remarkable above 13 repeat units, suggesting, as pointed out by Weber (1990), that informativeness tends to increase with the number of repeats.
The interruption of the perfect repeat array had a dramatic effect on the stability of the DQCAR alleles. It has been shown that imperfect repeats are less polymorphic (Weber 1990; Garza et al. 1995), and, interestingly, stabilization of repetitive sequences has also been described for certain trinucleotide repeats associated with diseases. In spinocerebellar ataxia type I (SCA1), Chung et al. (1993) showed that expanded alleles had a continuous run of (CAG)nrepeats in the SCA1 region, whereas unexpanded alleles had CAT interruptions. The gene for spinocerebellar ataxia 2 (SCA2) has been identified recently and also contains a stretch of (CAG)n repeats uninterrupted in mutated alleles and interrupted by one to three CAAs in normal ones (Imbert et al. 1996). Similarly, fragile X syndrome is associated with an expansion of an almost uninterrupted run of triple repeat (CGG)n at the 5′-untranslated region of the FMR1 gene with stable alleles showing interspersed AGGs (Eichler et al. 1994; Zhong et al. 1995).
It is not totally understood how interruptions stabilize repetitive sequences. Interruptions may create more matching points during replication, and a misalignment could be detected more easily by mismatch repair enzymes, as suggested by Heale and Petes (1995). They observed that in yeast, the interruption of a repetitive run by one different repeat increased by 100 times its stability, and this stabilization was dependent on a functional DNA mismatch repair system.
Interestingly, the phylogeny of DQB1/DQCAR haplotypes suggests that the C → A mutation interrupting the CA repeat is monophyletic and therefore occurred only once in the evolution of this locus. The interruption of the CA repeat array inDQ1-associated DQCAR alleles must have been present before the divergence of this subtype, as it was found in allDQCAR alleles associated with DQ1 haplotypes and in none of the non-DQ1 haplotypes. The four major DQ subfamilies (DQ1, DQ2, DQ3, and DQ4) can be identified in nonhuman primates. They have persisted for at least 5 million years, and diversity within the subfamilies seems to have arisen after speciation (Gyllensten et al. 1990).
Despite the low level of size variations in the DQCAR alleles associated with DQ1 haplotypes, additional sequence diversity was observed between alleles of identical size because of CA → GA changes at the end of the repeat. As the number of CA units increases, a concomitant decrease in the number of GA units keeps the overall allele size constant. Additional polymorphism is thus obtained for alleles of similar size, as observed previously at other microsatellite loci (Saha et al. 1993; Estoup et al. 1995). Sequence variation has also been observed in minisatellites (Jeffreys et al. 1994). The variation in the repetitive sequence is not restricted to DQCAR alleles associated with DQ1alleles but is also found in alleles associated with non-DQ1haplotypes (e.g., DQB1*0301 and DQB1*0402). Nevertheless, the frequent change of allelic composition of CA and GA repeat numbers in alleles of identical size suggests a new mutational mechanism that has not been reported previously in microsatellites.
In conclusion, sequence analysis shows a complex mutational pattern in a single (CA)n microsatellite. This complexity could also occur in other microsatellite loci and must be considered when using these markers for evolutionary and gene mapping studies.
METHODS
Tree Construction
A phylogeny of 14 frequent DQB1 alleles was reconstructed on the basis of the aligned coding sequences of these alleles using the neighbor-joining method (Saitou and Nei 1987). Genetic distances were estimated using Kimura’s two-parameter model (Kimura 1980). Several other measures of genetic distances were used, and all gave identical results in terms of the topology of the phylogeny obtained using the neighbor-joining method (Jin et al. 1996). The maximum parsimony tree displayed a slightly different topology, which became identical with the neighbor-joining tree once peptide-binding sites were removed from the analysis. The tree was rooted by DQB sequences of pig and horse. A maximum parsimony tree ofDQCAR locus based on the flanking sequences of DQCARalleles was also constructed.
Microsatellite Amplification and Sequence
Genomic DNA (200 ng) was used as a template, in a total volume of 25 μl of 10 mm Tris-HCl, 50 mm KCl, 1.5 mm MgCl2, 50 pmoles of each primer (see below), 125 μm of each dNTP, and 1 unit of AmpliTaq DNA polymerase (Perkin Elmer). After an initial incubation for 5 min at 95°C, the amplification was carried for 30 cycles, with each cycle consisting of 30 sec at 94°C, 1 min at 57°C, and 1 min at 72°C, with a final extension at 72°C for 15 min. The primers used were CAR1, 5′-GAAACATATATTAACAGAGACAGACAAA-3′; and CAR2, 5′-CATTTCTCTTCCTTATCACTTCATA-3′ (Satyanarayana and Strominger 1992; Macaubas et al. 1995). The presence of PCR product was confirmed by gel electrophoresis, followed by extraction with chloroform and ligation to pT7Blue T-vector (Novagen), as detailed in the product manual. Both strands of plasmid DNA from several clones were sequenced with Sequenase version 2.0 T7 DNA Polymerase (U.S. Biochemical), using T7 and U19 primers, following the manufacturer’s instructions
Cell Lines
Twenty HLA class II homozygous B cell lines were analyzed. Eighteen B cell lines were obtained from the Tenth Histocompatibility Workshop panel (Yang et al. 1987; Kimura et al. 1992; Aldener and Olerup 1993). The other two B cell lines were cell line MANN (donated by Dr. George Blanck, Harvard University, Cambridge, MA; Blanck and Strominger 1988) and B cell line no. 106, acquired from the UCLA DNA reference panel (Dr. P.I. Terasaki, University of California, Los Angeles).
HLA Typing
Chromosomes (2140) from several populations were typed forHLA–DQB1, HLA–DQA1 and DQCAR loci, and haplotypes were deduced as described in Kimura et al. (1992) and Jin et al. (1996).
Acknowledgments
This work was funded by National Institutes of Health (NIH) grants NS23724 and 33797 (to E.M.) and by NIH training grant T32-GS08404 (to L.J.) We thank Drs. P. Underhill and P. Oefner for suggestions regarding cloning and sequencing strategies, and Professor Jonathan Majer for reviewing the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Graylands/UWA Centre for Clinical Research in Neuropsychiatry, Gascoyne House, Graylands Hospital, WA 6010, Australia.
-
↵5 Corresponding author. Present address: TVW Telethon Institute for Child Health Research, West Perth, WA 6872, Australia.
-
E-MAIL macaubas{at}ichr.uwa.edu.au; FAX 61-9-388-3414.
-
- Received October 7, 1996.
- Accepted April 4, 1997.
- Cold Spring Harbor Laboratory Press








