
Man to Mouse—Lessons Learned from the Distal End of the Human X Chromosome
Conservation between human and murine X chromosomes has been accepted as an incontrovertible principle for a long time and was generally envisaged as a powerful model system for the evolution of vertebrate sex chromosomes. In fact, the original postulate of Ohno (1973) that the constraints imposed by X-inactivation would conserve genes on the mammalian X chromosomes was reevaluated to become a fundamental law underlying sex chromosome evolution.
Contrasting this overall picture of harmony, the most distal band of the human X chromosome (Xp22.3) reveals a unique and so far unprecedented picture of chromosomal rearrangements during the evolution in eutherian mammals. The result of efforts toward the mapping of murine homologs of human X chromosomal genes have not only illustrated the massive reshuffling of X chromosomes during their evolution but also refined our understanding of mouse models for certain X chromosomal human genetic disorders.
Human X Chromosome Genes with Murine Homologs Autosomal or Absent
Clearly the genes residing in the pseudoautosomal region (PAR1) of the human sex chromosomes are exceptional in their relative chromosomal localization in man and mouse. In 1992, Disteche et al. reported the first instance of a human pseudoautosomal gene with an autosomal localization in mouse. The gene for the α subunit of the granulocyte macrophage colony stimulating factor receptor (CSF2RA) resides on chromosome 19 in mouse (Disteche et al. 1992). This case indicated for the first time an incomplete conservation between the mouse and human X chromosomes and suggested that the genetic composition of the pseudoautosomal region may vary among eutherian mammals, probably because of chromosomal rearrangements. Disteche’s initial assumption was later confirmed by mapping additional murine homologs of human pseudoautosomal genes to mouse autosomes. The α chain of the heterodimeric interleukin-3 receptor (IL3RA) is encoded by a gene closely linked to the CSF2A receptor locus inPAR1 in humans (Kremer et al. 1993), yet its murine homolog maps to chromosome 14 in mouse (Milatowich et al. 1993).
ANT3, a gene encoding an adenine nucleotide translocating enzyme, was recently localized within PAR1 in humans and resides in direct vicinity to the cytokine receptor gene cluster discussed above (Schiebel et al. 1993; Slim et al. 1993). This gene could not be detected in the mouse genome (Ellison et al. 1996) and was therefore probably added to the human sex chromosomes during the 100 million years of evolution separating man and mouse.
The very latest example of a human pseudoautosomal gene with a distinct chromosomal localization in mouse not only revalidates once more the exceptional evolutionary position of these genes in general but also forces us to reconsider the significance of murine animal models for certain X chromosomal inherited disorders observed in humans. This gene, SHOX (or PHOG; Ellison et al. 1997), which encodes a homeodomain-containing protein, was recently identified by applying a positional cloning strategy aimed at a locus involved in the short stature phenotype in Turner patients (Rao et al. 1997). The putative murine homolog of SHOX (OG-12) was identified independently around the same time (Rovescalli et al. 1996) and maps to chromosome 3 in mouse (Ellison et al. 1997) (Fig.1). It is exactly this autosomal localization ofOG-12 that may explain the inadequate ability of the mouse model system (39,X0) to accurately resemble the short stature phenotype consistently observed in Turner syndrome in humans.
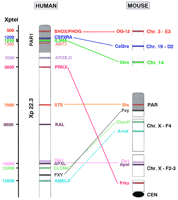
Eleven human genes residing within the most distal band of the human X chromosome (Xp22.3, left) are comparatively mapped in mouse, so far. The approximate physical distances of the human genes from the telomere (Xptel) are given at left. None of the genes localized in the human pseudoautosomal region (PAR1, shown in gray) are linked to the sex chromosomes in mice. The genes from within the X-specific portion of Xp22.3 are spread over the entire murine X chromosome. Mouse homologs for the human ANT3, ARSE, D, andKAL genes have not been identified. The asterisk denotes the different localization of Clcn4 in M. spretus andM. musculus.
Some have proposed that PAR1 originates from multiple additions and attritions from different autosomes on and off the sex chromosomes (Graves 1995). Taken together, the examples discussed so far clearly illustrate this assembly of the human pseudoautosomal region from a variety of different chromosomal loci during mammalian evolution.
Human X Chromosome Genes with Altered Murine Homolog X Chromosome Localization
Within the X-specific portion of Xp22.3, seven genes have been mapped comparatively in man and mouse. These seven genes reside within a stretch of DNA spanning <10,000 kb in humans while spreading over the entire 150,000 kb of the murine X chromosome (Fig. 1). The humanPRKX gene, encoding a putative serine/threonine protein kinase, maps proximal to the pseudoautosomal boundary on the X chromosome (Klink et al. 1995) and has a Y homolog located in Yp11.2 (Schiebel et al. 1997). In contrast, the murine Prkx gene was assigned to a region close to the X-chromosomal centromere in bothMus musculus and Mus spretus, thus residing at the almost maximal distance from the pseudoautosomal region on the mouse X chromosome (R.J. Blaschke, A. Mertz, S. Brown, and A. Rappold, in prep.). In addition, no Y homolog could be detected in either of these mouse species. The X-linked recessive steroid sulfatase (STS) gene, which causes the development of ichthyosis, was cloned in 1987 and mapped within the X-specific part of Xp22.3 (Yen et al. 1987). The murine homolog (Sts) shown previously to exhibit a segregation pattern consistent with an autosomal localization (Erikson et al. 1983;Keinanen et al. 1983) could in fact be localized within the pseudoautosomal region in mice (Salido et al. 1996) (Fig. 1).
Another gene cluster, consisting of the genes for ocular albinism type 1 (OA1) (Bassi et al. 1995), the apical protein Xenopus laevis-like (APXL) (Schiaffino et al. 1995), and a voltage gated chloride channel (CLCN4) (van Slegtenhorst et al. 1994), is located between STS and AMELX. Although closely linked in humans, these genes are well separated in mice (Fig. 1). An evolutionary breakpoint between Apxl and Clcn4separates the gene cluster with Oa1/Apxl remaining linked in mice. The murine Oa1 and Apxl genes reside within subinterval F2–F3 (Dinulos et al. 1996), whereas Clcn4 maps to subinterval F4 on the X chromosome in the wild Mediterranean mouseM. spretus but to chromosome 7 in laboratory strains ofM. musculus (Palmer et al. 1995; Rugarli et al. 1995). So far, this autosomal localization of Clcn4 in a laboratory mouse strain while another mouse strain carries it on the sex chromosome provides the only example of a murine gene contradicting Ohno’s law.
The FXY gene, which seems to be closely related to the gene responsible for the Opitz G/BBB syndrome (Quaderi et al. 1997), resides proximal to this gene cluster in humans (Perry et al. 1997), whereas its murine homolog Fxy crosses the pseudoautosomal boundary of the X chromosomes of mice but is truncated at the pseudoautosomal boundary of the Y chromosomes (Palmer et al. 1997). This truncation of the Y copy of the murine Fxy can be interpreted as an intermediate stage of the attrition process at work.
Finally, the most proximal gene taken into consideration here encodes the human enamel protein (Salido et al. 1992), which is involved in tooth development. This gene is well separated from the pseudoautosomal boundary in humans (Herell et al. 1995). In contrast, the murine homolog Amelx (Chapman et al. 1991) is located distal ofOa1/Apxl close to the pseudoautosomal region (Dinulos et al. 1996).
Summary
Looking over these mapping data, it is evident that the comparative mapping of Xp22.3 loci extends the recently suggested model for the evolution of mammalian X chromosomes by Blair et al. (1994). Rather than supporting the assumption of eight conserved X chromosomal regions being rearranged during mammalian evolution, data on Xp22.3 loci implicate a much more complex sequence of events leading to the divergence of the X chromosomes between man and mouse.
As more detailed knowledge of comparative maps becomes available, additional layers of rearrangement have been and are increasingly being revealed, and this is true of both the human versus mouse X chromosome comparison and that of the autosomes (DeBry and Seldin 1996). However, a comparison of the conserved linkage groups on the X chromosome to those on the autosomes once more exemplifies the unique situation on the chromosomal band Xp22.3. On autosomes, the number of mouse–human linkage groups range between 1 and 13 per chromosome. On the autosome with the highest number of conserved linkage groups, chromosome 7, a total of 13 linkage groups are spread over the entire 160-Mbp chromosome. In contrast, at least nine linkage groups are clustered on the distal 12 Mbp of DNA on Xp22.3. (For additional information on human–mouse comparative maps, see Carver and Stubbs, this issue).
Taken together, the recent comparative mapping data for genes within the most distal band of the human X chromosome illustrate the extremely eventful history of Xp22.3, emphasizing its unique evolutionary situation.
Having given a deeper meaning to this extraordinary situation of Xp22.3 should allow us to push ahead the investigation of this region in at least two ways. On the one hand, the extension of our knowledge about the comparative localization of genes residing within Xp22.3 facilitates the development of new and more sophisticated model systems for the evolution of vertebrate sex chromosomes in general and permits the characterization of additional breakpoints leading to the chromosomal rearrangements between man and mouse. Clearly, such considerations lend more interest to evolutionary comparisons beyond the mouse, putting it in the wider context of mammalian evolution (Table 1). These latest data could stimulate the development of new and more specialized models no longer based on the overall conservation of the vertebrate sex chromosomes but, rather, paying tribute to the exceptional genetic instability of Xp22.3. In stressing the difference instead of invoking the similarity, such new model systems could be extremely helpful to our understanding of the mechanisms underlying the chromosomal instability of this region.
Comparative Mapping Position of Human Xp22.3 Loci in a Variety of Different Mammalian Species
After all, it might be just the same principles driving the massive evolutionary rearrangements that also account for the accumulation of deletions and/or translocations affecting this chromosomal region, thereby leading to the occurence of individual genetic disorders or combinations thereof, the so-called “contiguous gene syndromes” (Ballabio and Andria 1992).
Acknowledgments
We thank Steve Brown (Harwell) and Jennifer A.M. Graves (Melbourne) for their comments on the manuscript.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL gudrun_rappold{at}krzmail.krz.uni-heidelberg.de; FAX 06221/56-5332.
- Cold Spring Harbor Laboratory Press








