
Sequence Ready—or Not?
En route to the goal of the Human Genome Project (HGP) of obtaining the complete sequence of the human genome by the year 2005, several milestones have been reached (Collins and Galas 1993). First was the construction of a genetic linkage map with an average resolution of 1 cM (Dib et al. 1996). Second was the construction of a physical map with an average marker spacing of <150 kb (Hudson et al. 1995). These are laudable achievements; however, they only represent the completion of the initial phase of generating a sequence-ready physical map of the human genome that will provide the necessary templates for large-scale sequencing.
The current physical map is composed of two main resources. The first is physical maps made up of overlapping cloned fragments of human DNA. These clone contigs have been largely constructed using sequence-tagged site (STS) content mapping methods (Green and Olson 1990). The most comprehensive human genome physical maps are largely composed of overlapping yeast artificial chromosome [(YAC) Burke et al. 1992] clones [see http://www-genome.wi.mit.edu (Cohen et al. 1993; Chumakov et al. 1995; Hudson et al. 1995)]. Unfortunately, YAC clones are not suitable substrates for large-scale sequencing of the human genome. This is largely due to the high rate of chimaerism, the high frequency of deletions and rearrangements observed in these clones, and the difficulty of obtaining purified YAC DNA. The second physical map resource that is widely used is the whole-genome radiation hybrid (RH) map (Hudson et al. 1995; Schuler et al. 1996;Stewart et al. 1997). This map provides many additional ordered and binned markers and has been integrated with the YAC map. It is important to realize that although the average STS spacing is approximately one every 150 kb, the distribution is not random. A large number of these markers have been derived for positional cloning projects or from expressed sequence tags (ESTs), which may enrich marker density in disease regions or gene-rich regions, respectively. The combined maps provide much of the basis for the current effort to generate a sequence-ready map of the human genome.
A sequence-ready map must meet certain criteria. Obviously, it must be composed of clones that are suitable sequence templates. Experience with sequencing the yeast and Caenorhabditis elegans genomes has shown that bacterial clones are an excellent choice. The primary clones being used are bacterial artificial chromosomes [(BACs) Shizuya et al. 1992], P1-derived artificial chromosomes [(PACs) Ioannou et al. 1994], and cosmids (Wahl et al. 1987). The clones corresponding to the STSs from a target region are typically isolated from arrayed libraries using PCR or hybridization strategies (see Fig.1). An alternate approach is to subclone YAC clones into cosmids directly (Wong et al. 1997). Once the appropriate clones have been identified, the next step is to assemble these clones into an ordered contig. Clones are subjected to restriction endonuclease digestion, and fragments are separated by gel electrophoresis (Marra et al. 1997; Wong et al. 1997). Overlapping clones are then identified by shared restriction pattern or “fingerprint” similarities (see Fig. 2). It is a critical requirement for a sequence-ready map that the contigs have sufficient depth so that all restriction fragments can be validated by several overlapping clones. That is, if restriction fragment inconsistencies are observed, enough clones must be examined to establish the correct fragment pattern from the anomalous one. This is further complicated by the presence of polymorphisms. The BAC and PAC libraries being used for assembling sequence-ready maps of the human genome have two, and often more, haplotypes. Experience has shown that 6- to 10-fold coverage is usually acceptable. It may be necessary to screen several libraries to achieve this depth uniformly across a genomic segment. It is only through this validation that clone integrity can be assured. The desired sequence error rate of not more than 1 error in 10,000 bp that has been set for the human genome is meaningless if deleted or rearranged clones are selected for sequencing. In addition, increased clone depth in a contig allows for the selection of efficient tiling paths. The desired tiling path is the set of overlapping validated clones that represent the genomic region to be sequenced while maintaining a minimal overlap between any two clones. Most large-scale sequencing centers employ a shotgun sequencing strategy. This involves randomly fragmenting the clone to be sequenced into smaller fragments (1–1.5 kb), subcloning these fragments into a suitable vector and sequencing sufficient subclones to achieve approximately a sixfold coverage of the original clone (Wilson and Mardis 1997). This strategy necessitates the redundant sequencing of a region of overlap between two adjacent clones in the minimal tiling path. Obviously, smaller overlaps represent cost-savings in this process. It needs to be stressed that this only applies to the initial random shotgun phase of the project, as overlap regions are only finished to the 1 error in 10,000 gold standard for one of the clones.
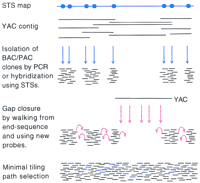
Sequence-ready map generation.

Verification of clone integrity by fingerprint analysis. Shown is a representation of the restriction patterns of seven overlapping clones in a contig. Each lane (A–G) represents a single clone. Vector-specific fragments are shown in green. All fragments in clone D (shown in blue) are verified by their presence in its overlapping neighbors (red lines).
A related issue to minimal overlap is contiguity of large regions of the genome in advance of the selection of the minimal tiling path. To achieve the complete sequence of the genome, all contigs must be joined to their neighboring contig with the same requirements needed for the original contigs as described above. Two strategies for gap closure are shown in Figure 1, namely end-walking from selected clones in a contig and the generation of new probes within a gap using the overlying YAC clones. Selecting clones for sequencing from small contigs leads to a large number of gaps that will need to be closed. Closing small gaps with large clones leads to excessive overlaps and eliminates the ability to select an efficient minimal tiling path. This is one of the biggest challenges facing the preparation of a sequence-ready map of the human genome. The state of the map at the start of the sequencing phase of the HGP and the rate of sequencing scale-up needed to complete the task by the year 2005 make achieving contiguity difficult while providing sufficient template at the rate needed by a large center. Much progress has been made in large-scale sequence-ready mapping in the past year. New and more efficient strategies are being developed that will allow physical mapping to advance beyond the immediate urgent need for validated clones such that contiguity can be achieved.
In summary, a sequence-ready map must meet the following criteria. (1) It must be composed of clones that are efficient sequencing templates. (2) It must be assembled using a method that examines the entire cloned insert such as restriction digest fingerprinting. Simply using STS content does not allow the determination of the extent of overlap of any two clones nor does it detect deletions and rearrangements not involving the STSs. (3) The contig must have sufficient depth to allow the validation of the selected clones in the face of restriction pattern inconsistencies. In addition, increased contig depth will allow the selection of a more efficient minimal tiling path. A uniform depth of 6- to 10-fold coverage is recommended. (4) For large-scale projects, as large a contig as possible should be constructed in advance of selecting the minimal tiling path to avoid future closure problems. Contigs of at least 1 Mb are desirable, albeit difficult and time-consuming to achieve.
Except for a few select segments, the current human genome maps do not meet many of the criteria discussed above. The ultimate quality of the human genome sequence that is being obtained is as much dependent on the conversion of these maps to a sequence-ready product as it is on the accuracy of the sequencing itself.
Acknowledgments
Thanks go to Elaine Mardis and Marco Marra for valuable discussions regarding this paper.
Footnotes
-
↵1 E-MAIL ; FAX (314) 286-1810.
- Cold Spring Harbor Laboratory Press








