
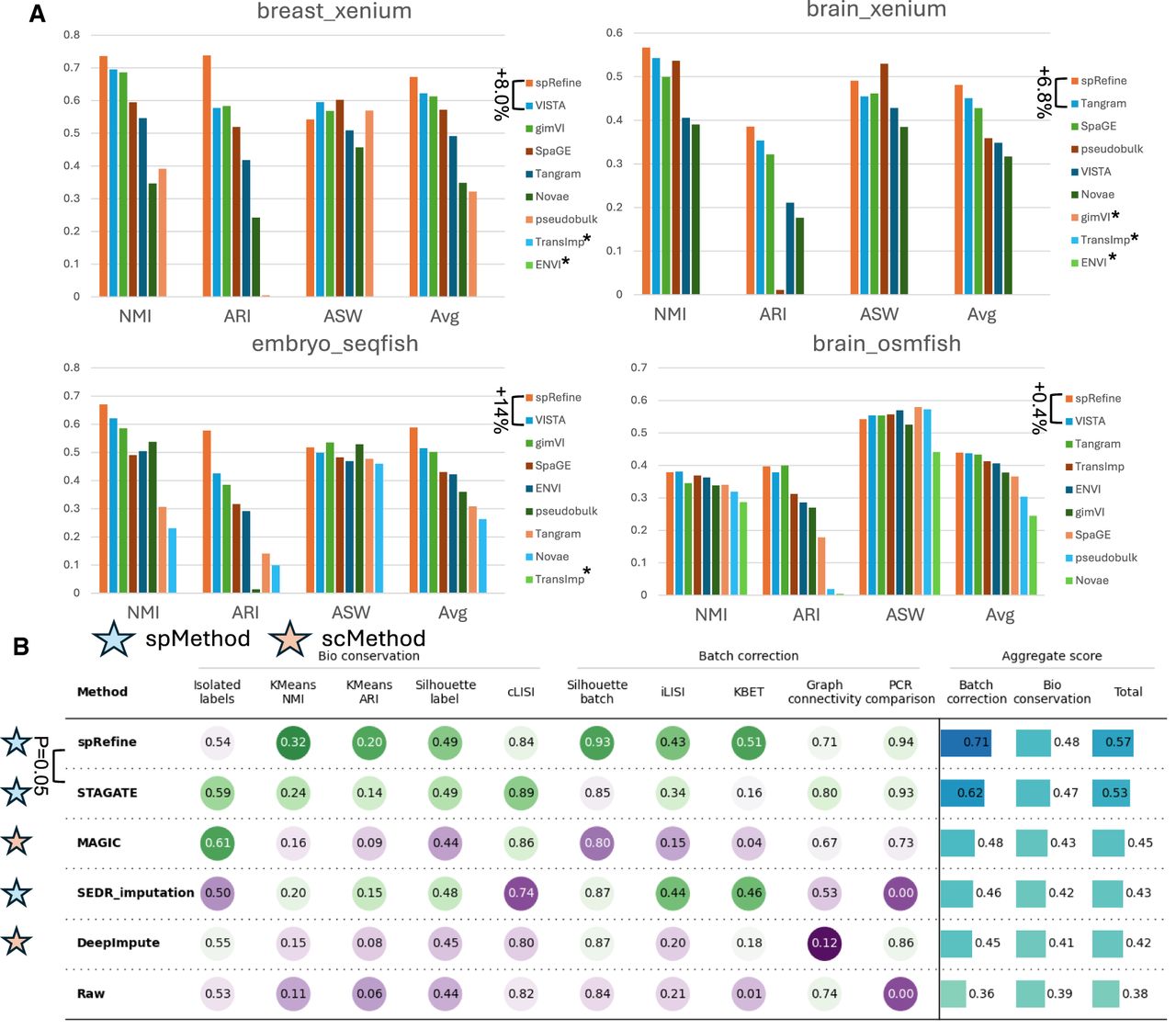
Comparisons between spRefine and other baselines for cell clustering and batch effect correction. (A) The clustering performance based on different imputation methods or spatial foundation models across four sub-cellular-resolved spatial transcriptomic data. The star means that the selected method met out-of-memory (OOM) errors in the specific data set. The methods are ranked by the Avg score for each data set, and we highlight the increment made by spRefine compared with the second-best baseline method. (B) The performance of batch effect correction based on different methods for processing the SpatialLIBD data set. Here, spMethod represents the method designed for spatial transcriptomics, whereas scMethod represents the method designed for single-cell transcriptomics. We performed the Wilcoxon rank-sum test (one-side) between spRefine and the second-best baseline method, and annotated the p-value (P) in this panel.








