
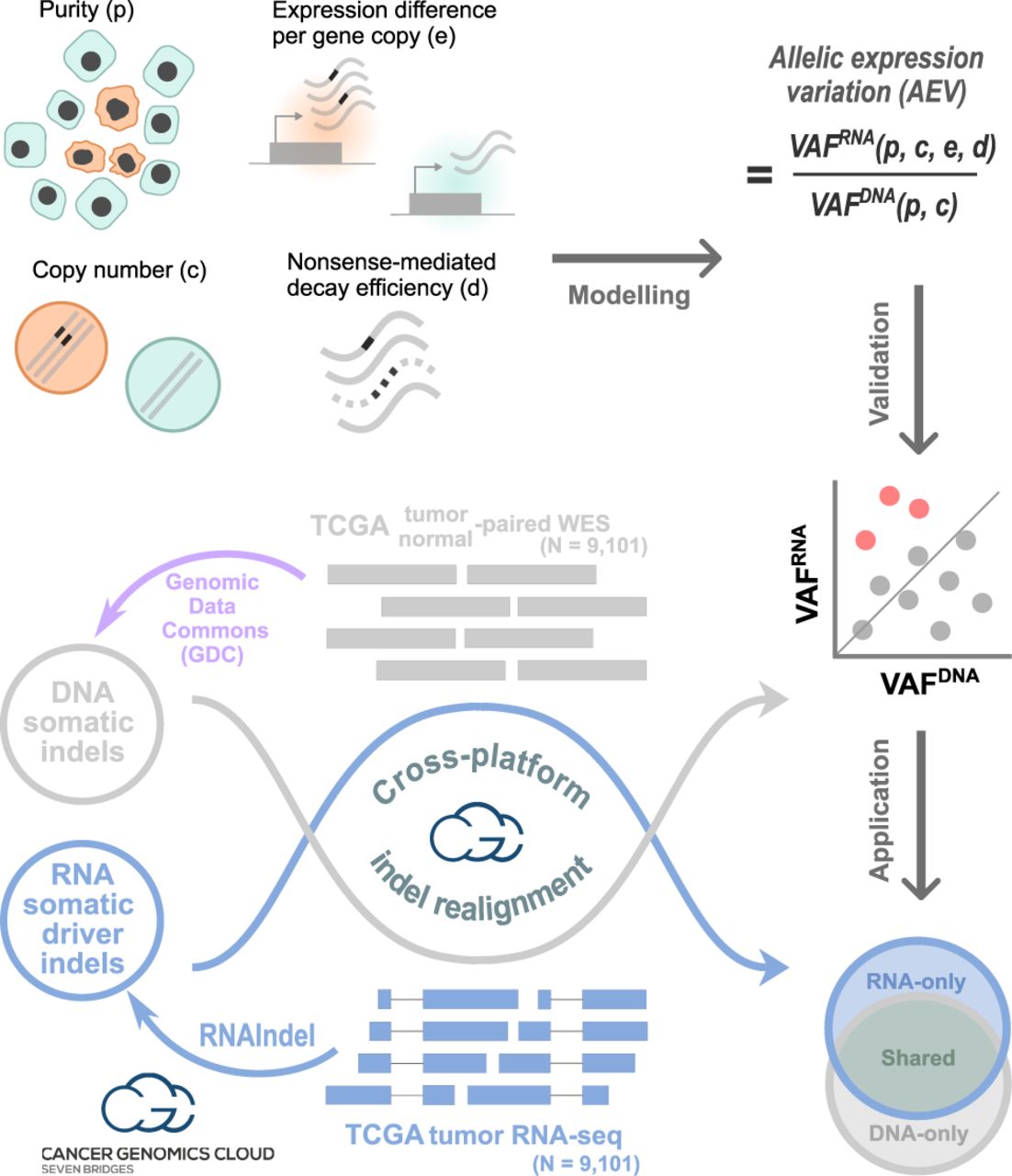
Study design with three main workflows. In modeling, the purity, copy number, expression level per copy in tumor relative to normal, and nonsense-mediated decay (NMD) efficiency were used to model the VAF ratio of RNA to DNA (i.e., VAFRNA/VAFDNA), which is termed allelic expression variation (AEV). Theoretical and simulation studies were conducted to identify conditions for AEV > 1 (elevated AEV). In validation, DNA somatic indels identified by the Genomic Data Commons's (GDC) multicaller platform in the TCGA data set (purple workflow) were used to validate the model for elevated AEV (red dots). Reads supporting indels were realigned in both WES and RNA-seq to accurately calculate AEV. In application, confounding effects leading to elevated AEV can be exploited to enhance mutation calling from RNA-seq. This was tested for somatic driver indel detection by calling from RNA-seq with WES read support to complement the TCGA indel set (Venn diagram). Indels in each Venn section were examined for AEV and purity along with potential clinical implications. Major computational analyses for validation and application were performed on the Cancer Genomics Cloud.








