
Tissular chromatin-state cartography based on double-barcoded DNA arrays that capture unloaded PA-Tn5 transposase
Abstract
Recent developments in spatial omics are revolutionizing our understanding of tissue structure organization and their deregulation in disease. Here, we present a strategy for capturing chromatin histone modification signatures across tissue sections by taking advantage of a double-barcoded DNA arrays design compatible with in situ Protein A–transposase Tn5 tagmentation. This approach has been validated in presence of fresh-frozen mouse brain tissues but also in decalcified formalin-fixed paraffin-embedded (FFPE) mouse paw samples, in which either the histone modification H3K4 trimethylation or H3K27 acetylation has been used as proxy for interrogating active promoter signatures. Furthermore, because combinatorial enrichment of multiple histone modifications was shown to code for various states of gene transcriptional status (active, bivalent, repressed), we have integrated several histone modifications generated from consecutive mouse embryo sections to reveal changes in chromatin states across the tissue. Overall, this spatial epigenomic technology combined with the use of a spatial chromatin-state analytical strategy paves the way for future epigenetics studies for addressing tissue architecture complexity.
Developments in spatially resolved transcriptomics (SrT), and more recently on spatially resolved epigenomics (Deng et al. 2022), are providing the means to interrogate organ/tissue architecture from the angle of the gene programs defining their molecular complexity. Despite the proven performance of the available spatially resolved omics technologies in a variety of tissue samples; there are still major challenges to address. Indeed, beyond the overdiscussed aspects related to the resolution of these technologies, their capacity to access to other type of molecular readouts (e.g., chromatin accessibility, epigenetics), their technical limitations resulting from the way in which the biological specimens were preserved (formalin-fixed paraffin-embedded [FFPE] vs. fresh-frozen tissues), and their current elevated costs make these technologies applicable to few tissue sections, thus avoiding generating multiomic integrative views of their molecular complexity.
Previously, we have presented a double-barcoded DNA array strategy for SrT, allowing us to interrogate large numbers of tissue sections in a cost-efficient manner, as illustrated by the setup of a three-dimensional molecular cartography of human cerebral organoids (Lozachmeur et al. 2023). Herein, we present the use of double-barcoded DNA arrays to perform spatial epigenomic landscaping. Specifically, this implies (1) a modified version of double-barcoded DNA arrays for capturing cleaved chromatin with the CUT&Tag chemistry (Kaya-Okur et al. 2019); (2) a modified bioinformatics pipeline allowing us to demultiplex spatial read-counts retrieved around gene promoter regions and their visualization within our tool MULTILAYER (Moehlin et al. 2021b); and (3) the implementation of combinatorial epigenetic enrichment analysis generated from consecutive sections, allowing us to reveal chromatin states associated to gene promoters.
Our double-barcoded spatial epigenomic strategy has been validated in fresh-frozen tissues but also in decalcified FFPE conserved material (bone-related tissues), known to be incompatible with spatial transcriptomic assays because of RNA degradation. Finally, this technology has been used for performing spatial epigenomic profiling in mouse embryo samples, targeting several histone modifications assessed in consecutive sections. This strategy allows the revealing of relevant chromatin-state signatures associated to either active, repressive, or bivalent chromatin status across the tissue, thus providing a detailed molecular view of the chromatin regulation on the grounds of its physical location.
Overall, this study covers both the development of a cost-efficient strategy for generating spatial epigenomic maps on tissue sections and a bioinformatics strategy for a pioneer integrative effort in spatial chromatin-state profiling, essential for the field of “spatial omics,” anticipating the need for this type of effort with the democratization of these technologies.
Results
A double-barcoded spatial epigenomic strategy allowing the capture of histone modification signatures across large tissue sections
With the aim of capturing histone modifications across tissue sections, we have engineered a DNA array manufacturing strategy based on the use of two complementary oligonucleotides printed at a high accuracy with the help of the picoliter spotter sciFLEXARRAYER S3 (Scienion). As in our previous design for double-barcoded spatial transcriptomics (Lozachmeur et al. 2023), we have first deposited, as rows, the oligonucleotides harboring an amino C6 linker modification at the 5′-end for UV cross-linking, a T7 promoter sequence, a unique molecular barcode per deposited row, and a 30-nt-long adapter sequence, herein labeled as “Gibson” (Fig. 1A). Then, we have printed per column and on top of the previous deposited probes, the second set of oligonucleotides harboring a complementary Gibson sequence at the 3′-end, a unique molecular barcode per deposited column, and a specific 19 nt transposase recognition sequence (mosaic end [MOS]).
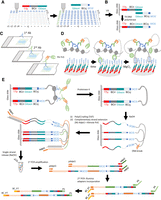
Principle behind the double-barcoded spatial epigenomic technology. (A) DNA arrays are printed by depositing first probes in rows, followed by deposition of a second type of probes in columns. The first type of probe is composed of a T7 promoter (T7p) sequence, a unique molecular barcode associated to the row (BCr1, …BCri), and bridge sequence called “Gibson.” The second type of probe presents a complementary Gibson sequence, a unique molecular barcode associated to the column (BCc1, …BCcj), and a complementary mosaic sequence (MOS′). (B) Because both probes were combined during printing, after UV irradiation for immobilization, hybridized probes are elongated with the T4 DNA polymerase, leading to a double-strand molecule presenting the mosaic sequence (MOS) at the free end of the probe. (C) After depositing a tissue section on top of the manufactured DNA array, a first antibody against the protein of interest is incubated, followed by a second antibody against the first antibody, and is finally incubated with the recombinant Protein A–transposase PA-Tn5. (D) Scheme illustrating the loading of the Tn5 into the mosaic sequence retrieved on the printed probes on the DNA array, followed by the chromatin cleavage induced by magnesium chloride, leading to the formation of hairpin-like DNA structures. (E) Scheme illustrating the molecular biology steps required for generating a copy of the genomic DNA captured by the DNA probes, followed by their amplification for Illumina massive parallel DNA sequencing.
Printed DNA arrays are first UV-irradiated to cross-link the DNA probes, and then they are covered with a solution containing T4 DNA polymerase for elongating the hybridized probes, leading to a double-stranded DNA molecule harboring a unique combination of two molecular barcodes, as well as a MOS sequence at the exposed extremity of the DNA probe (Fig. 1B). This strategy allows the manufacture of DNA arrays composed by several thousands of DNA probes in a cost-effective manner because the molecular barcodes’ complexity is obtained in a combinatorial manner.
Tissue sections deposited on top of the double-barcoded DNA arrays are permeabilized and incubated with an antibody against the histone modification of interest, followed by a secondary antibody binding allowing the enhancement of the tethering of the Protein A–transposase Tn5 (PA-Tn5) (Fig. 1C). In contrast to the previously described spatial CUT&Tag strategy (Deng et al. 2022), herein we use a PA-Tn5 moiety devoid of loaded DNA sequences, such that the loading takes place with the MOS sequences retrieved in the exposed ends of the printed DNA probes (Fig. 1D). In that manner, the cleaved chromatin in presence of magnesium chloride remains captured by the proximal DNA probes (Fig. 1E). After tissue degradation by Proteinase K treatment, the DNA array is exposed to alkaline conditions (NaOH), revealing single-DNA ends for library preparation in situ. This is performed by extending DNA extremities by poly(C) tailing with terminal transferase and hybridization of a poly(G) primer presenting a known adapter sequence at its 5′-extremity (Adpt1), followed by Klenow polymerase extension (Fig. 1E). Finally, DNA arrays are exposed again to alkaline conditions for releasing the elongated complementary DNA sequence and are collected in a tube for performing two rounds of PCR amplification. The first PCR amplification step relies on a primer complementary to the T7-promoter sequence and presenting a known adapter fragment (Adpt2) as well as on a primer targeting the Adpt1. The second PCR amplification uses the Adpt1 and Adpt2 sequences for introducing the P5 and P7 Illumina adapters, including a molecular barcode sequence specific to the prepared library (Fig. 1E).
This strategy has been validated in an adult mouse brain fresh-frozen tissue section deposited on top of a DNA array covering a surface of 16 × 8 mm, presenting a total of 4096 unique combinations of dual barcodes (Fig. 2A). The manufactured DNA array presented a pitch distance of about 177 microns, with a DNA probe resolution of 100 microns (diameter spot) (Fig. 2B). The tissue section has been immunolabeled with an antibody targeting the histone modification H3K4me3, known to be enriched in actively transcribing or bivalent promoter regions. For enhancing the tethering of the PA-Tn5, a secondary antibody has been added (1/100 dilution), complemented by the same secondary antibody conjugated to a fluorophore (AF-488) but loaded at a higher dilution level (1/1000). In that manner, the performance of the histone modification labeling can be visualized in the same sample that will be used for the spatial epigenomic profiling (Fig. 2C).
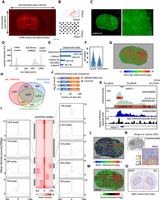
Spatial epigenomic profiling over a whole-mouse brain section. (A) Micrograph displaying the scanning of a mouse brain coronal section deposited on top of a DNA array composed of 64 × 32 interstitial printed probes harboring the mosaic sequence. Three rows of Cy3-labeled probes are visible at the border of the micrograph, corresponding to fiducials used for defining the physical position of the tissue section across the 4096 printed probes. Notice that the DNA array covers a surface of 16 × 8 mm. (B) Micrograph displaying a bright-field scanning of a DNA array printed area in which each of the printed probes is visible as a result of salt deposition. Due to the interstitial printing, the pitch distance resolution of the array is ∼177 microns. (C) Immunolabeling of the section displayed in A, revealing the histone modification H3K4me3. The inset magnification area reveals the nuclear staining visible through this labeling, as well as the differences in cell density across different parts of the mouse brain section. (D) Electropherogram displaying the spatial epigenomic (SpE) Illumina sequencing library obtained from the mouse brain section. (E) Number of total sequenced reads, as well as those recovered at different stages of the primary bioinformatics analysis. (F) Violin plots displaying the number of read counts or promoter regions per spatial epigenomic element (SpExel), in analogy to picture elements (pixels). (G) Digital display corresponding to the number of adjusted read counts associated to the physical positions in the mouse brain section (heatmap displayed in logarithmic scale). (H) Venn diagram displaying the number of promoters presenting H3K4me3 peaks in common between the SpE map and two bulk H3K4me3 ChIP-sequencing profiles (GSM1656749, GSM1000095). For this comparison, all mapped reads across the tissue were collapsed to generate a pseudobulk profile and processed with the peak caller MACS2 (P-value < 1 × 10−5). (I) Peak-centered heatmap for the H3K4me3 peaks retrieved in common between the SpE map and both bulk data sets. Insets display average peak density retrieved in nine peak classes generated from peak clustering based on their shape. (J) Common peaks retrieved between the spatial H3K4me3 profile and the spatial data published by Deng et al. (2022) (GSM5622964). Notice that more than 5000 to 8000 peaks are retrieved in common, on the grounds of the confidence threshold used for the comparison (MACS2 peak calling applied on the corresponding pseudobulk data). (K) Genome browser views assessed around the promoter region associated to the gene adenyl cyclase (Adcy5). (Top) Bulk ChIP-sequencing data from mouse brain samples and visualized within the QC Genomics genome browser. Heatmap on the bottom of each profile corresponds to a local QC enrichment assessment (Mendoza-Parra et al. 2013). (Bottom) Pseudobulk enrichment profiles issued either from the public data set generated by Deng et al. (2022) (GSM5622964; blue) or from the H3K4me3 enrichment map generated in this study (black). (L) Spatial H3K4me3 enrichment signatures retrieved in the promoter region associated to the gene Adcy5. (M) Same as L, but expressed in a differential enrichment context relative to the average levels across the tissue. (N) Mouse brain tissue section analyzed by Deng et al. (2022) and the corresponding H3K4me3 enrichment spatial signature associated to the gene Adcy5. Image adapted from Deng et al. (2022). (O) In situ hybridization (ISH) on a mouse brain section for the gene Adcy5 (Allen Mouse Brain Atlas).
Once the immunolabeled mouse brain section was scanned for revealing the histone modification localization (FITC channel) (Fig. 2C) and for capturing its physical position within the DNA array through Cy3-labeled fiducial oligonucleotides deposited in the borders of the double-barcoded deposited probes (TRITC channel) (Fig. 2A), the aforementioned procedure was applied, leading to a sequencing library presenting DNA fragments ranging between 300 and 1000 bp (Fig. 2D). More than 250 million reads were Illumina-sequenced, and a custom-made bioinformatics pipeline has been applied, targeting the detection of the Gibson sequence, the presence of both unique molecular barcodes, and the mapping to the mouse genome of the captured genomic sequence (Supplemental Fig. S1A; Fig. 2E). Despite the major loss of sequenced reads during this primary data processing, more than 40 million reads mapped to the mouse genome and were processed as bulk with the peak caller MACS2 (Zhang et al. 2008), followed by the identification of promoter regions presenting confident peaks. Demultiplexing of these confident regions gave rise to about 2000 read counts per spatial epigenomic element (SpExel; in analogy to pixel for picture element) and about 1500 promoters per SpExel in average (Fig. 2F).
Spatial epigenomic maps generated from confident peaks in promoters were first quantile-normalized for correcting the potential technical variations across printed probes and then adjusted for cell density variation across the tissue by integrating immunostaining pixel information with the assessed read count levels (Supplemental Fig. S1B). The obtained mouse brain spatial H3K4me3 epigenomic map revealed adjusted read counts fluctuating between 11 and 18 (log2 scale), in which the corpus callosum structure is visible owing to its high adjusted read count intensity (Fig. 2G). Promoters enriched for the H3K4me3 histone modification across the spatial map were compared with those described in two public bulk epigenomic maps assessed on mouse brain samples, revealing 8097 common genes, corresponding to >60% of all retrieved peaks in either condition (bulk or spatial setting) (Fig. 2H). Importantly, a peak-centered heatmap display of these common H3K4me3 peaks revealed preferentially the presence of a two-peak shape signature around the TSS, which has been further stratified in nine peak classes based on their intensity but also on their surrounding signatures (Fig. 2I). Indeed, beyond having a double-peak TSS-centered signature, H3K4me3 enrichment around the TSS can further expand in the direction of the active transcription, correlated with an increased transcription elongation (Chen et al. 2015). This has been further illustrated in the case of the gene Adcy5, known to encode for a member of the membrane-bound adenylyl cyclase enzymes and to be expressed in the brain. As illustrated in Figure 2K, the H3K4me3 enrichment around the promoter region of Adcy5 analyzed in the aforementioned ChIP-seq data sets, reveals a broader signature in one of the data sets (GSM1656749) and notably skewed toward the gene transcription orientation. Similarly, the pseudobulk profiles obtained either from a public H3K4me3 spatial data (GSM5622964) or from the one generated in this study present H3K4me3 enrichment at the level of the TSS of Adcy5 but also further enrichment signatures within the gene transcription orientation (Fig. 2K).
Comparing the pseudobulk profile obtained from a public H3K4me3 spatial data (GSM5622964) with the one obtained in this study revealed between 5000 and 8000 common peaks (depending on the enrichment confidence threshold in use), corresponding to >40% of the peaks retrieved in the public data, despite the few number of read counts used for the analysis (about 40 million reads after mapping) (Fig. 2J).
Finally, the spatial H3K4me3 enrichment signature associated with the gene Adcy5 has been compared either with the one generated by GSM5622964 (Deng et al. 2022) or with data provided by the Allen Mouse Brain Atlas. Indeed, Adcy5 has been previously shown to be overexpressed in the mouse brain basal ganglia (Fig. 2O), concordant with the spatial signature revealed in the obtained H3K4me3 map (Fig. 2L,M), as well as in the H3K4me3 spatial map published by Deng et al. (2022), despite the fact that these last published data cover a rather small part of the left lobe of the mouse brain section (Fig. 2n).
Similarly, H3K4me3 enrichment in the promoter region associated to the gene THAP domain-containing, apoptosis-associated protein 2 (Thap2), glucose 6 phosphatase, catalytic, 3 (G6pc3), or the transcription factor Sox10 presented spatial signatures mainly concentrated around the corpus callosum structure in agreement with the data provided in the H3K4me3 spatial map published by Zhang et al. (2023) (Supplemental Fig. S2A,B). Furthermore, pseudobulk profiles obtained either from the public H3K4me3 spatial data (GSM5622964) or the one generated in this study, revealed similar enrichment profiles around the promoter regions of the aforementioned genes; in coherence with bulk H3K4me3 profiles obtained from two public mouse brain data sets (Supplemental Fig. S2C). In the particular case of Sox10, in contrast to the data generated by Deng et al. (2022) (Supplemental Fig. S2D), our H3K4me3 spatial enrichment profile appeared extremely sparse, potentially owing to low sequencing coverage, but it could also be explained by low gene expression behavior, as revealed by in situ hybridization data from the Allen Mouse Brain Atlas (Supplemental Fig. S2E).
Overall, we have demonstrated herein that despite a low coverage used for the spatial profile reconstruction, our spatial epigenomic profiling allowed to capture relevant H3K4me3 enrichment signatures across a mouse brain section, in agreement with previous studies.
Major molecular players involved in rheumatoid arthritis can be detected by spatial epigenomic profiling performed in decalcified FFPE mouse paw samples
One of the major limitations of the available spatial transcriptomics technologies is their capacity to interrogate biological material in which the storage condition is not RNA friendly. Specifically, this corresponds to FFPE tissue samples, classically used for clinical sample preservation. Although current spatial transcriptomic methods interrogate these type of samples by targeting the remaining RNA by hybridization, this strategy reaches its limits in bone-related samples in which an acid-based decalcification procedure, prior FFPE embedding, further destroys the remaining RNA (Duncan et al. 2019).
Considering that acid-decalcified FFPE samples are classically used for immunostaining assays after deparaffination and rehydration, we thought that our spatial epigenomic strategy should be able to capture histone modification-enriched chromatin. To address the performance of our spatial epigenomic methodology in bone-related FFPE samples, we utilized samples from a collagen-induced arthritis (CIA) mouse model (Brand et al. 2007). Specifically, CIA mouse paws were fixed with paraformaldehyde and decalcified in the presence of formic acid and FFPE (Fig. 3A). FFPE blocks were cut to obtain thin sections that were deposited on top of our manufactured DNA arrays harboring mosaic sequences. DNA arrays were deparaffined and rehydrated by following standard procedures, followed by spatial epigenomic processing (Fig. 3A).
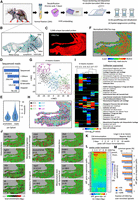
Spatial epigenomic assay in decalcified FFPE tissues. (A) Strategy for analyzing mice paw tissues from a collagen-induced arthritis (CIA) model. (B) Representative mouse paw tissue section revealing the presence of the tibia, talus, navicular, cuneiform, and metatarsus bone. (C) H3K27-acetylation (H3K27ac) spatial epigenomic profiling performed on a mouse paw tissue section deposited on a double-barcoded DNA array (64 × 32 printed probes). (D) Number of total sequenced reads and those recovered at different stages of the primary bioinformatics analysis. (E) Violin plots displaying the number of read counts or promoter regions per SpExel. (F) Digital view of the normalized and adjusted number of read counts associated to the physical location (SpExel) of the tissue section. (G) T-distributed stochastic neighbor embedding dimensionality reduction analysis (t-SNE) displaying the presence of six clusters of SpExel; (H) Clusters’ projection on top of the physical position within the tissue. (I) Cell type gene marker association analysis performed per clusters identified in G. Relevant cell types retrieved enriched per cluster are highlighted (green boxes). (J) Example of gene promoters found enriched in H3K27ac read counts and associated to the aforementioned clusters. Cell type annotations indicated are collected from the CellMarker augmented database used for this analysis. (K) Scheme of the CIA immunization assay performed by Li et al. (2022), allowing the collection of bulk-transcriptomics data from mice paw soft tissue at different time points. (L) Heatmap revealing differential gene expression signatures from the soft-tissue transcriptomes and classified in seven gene coexpression paths. Along the public transcriptome, the detection of H3K27ac overenrichment across the decalcified FFPE mouse paw section (number of overenriched SpExel) is displayed (blue heatmap). (M) Fraction of common genes between those associated to one of the gene coexpression paths in L and those presenting a H3K27ac enrichment in the spatial epigenomic map. Notice that only H3K27ac overenriched promoters (log-fold change [LFC]>1) being present in >1%, >5%, or >10% of total SpExel across the section were considered in this analysis.
Sagittal sectioning of the mouse paws allows evaluation of the ankle and tarsal joints, visualized by the presence of a sagittal plane of the talus, navicular, cuneiform, and metatarsus bones (Fig. 3B). To capture potential actively transcribing genes, we have performed a spatial epigenomic profiling targeting the histone modification H3K27 acetylation (H3K27ac) over a DNA array composed of 2048 unique double-barcoded DNA probes (Supplemental Fig. S3; Fig. 3C). Illumina sequencing of the obtained spatial epigenomic library provided more than 330 million reads, from which ∼77% presented the Gibson sequence, from which ∼40% presented dual barcodes (Fig. 3D). Mapping to the mouse reference genome allowed us to identify about 78 million reads, which were processed as bulk for identifying confident peaks within promoter regions. Demultiplexing of these confident regions gave rise to approximately 2000 read counts per SpExel and approximately 1800 promoters per SpExel in average (Fig. 3E).
A H3K27ac spatial epigenomic map generated from confident peaks in promoters was quantile-normalized and adjusted for variations in tissue density, leading to a digital map representing the adjusted read counts per physical position across the tissue (Supplemental Fig. S3; Fig. 3F). A T-distributed stochastic neighbor embedding (t-SNE) analysis stratified the corresponding SpExel in six clusters, which were then projected on the spatial tissue map (Fig. 3G,H). Cell type gene marker association analysis allowed the identification of major relevant terms corresponding to bone composition (mesenchymal progenitors, clusters 1 and 6; osteocytes, clusters 1, 2, and 3; osteoblasts, clusters 2 and 6), cartilage (cartilage progenitor cell, clusters 1 and 6; mesenchymal progenitors, cluster 6), as well as the synovial joint (mesenchymal stem cells: synovial fluid, cluster 4) (Fig. 3I). Furthermore, cell type gene markers associated to an inflammatory response were detected, including the presence of regulatory T cell signatures (FOXP3+ natural regulatory T [Treg] cells, monocytes, cluster 4 and 5; large granular lymphocytes, cluster 6; regulatory T cells, clusters 1, 4, 5, and 6) and markers associated with osteoarthritic cartilage tissue (mesenchymal progenitors, cluster 4) (Fig. 3I).
To further support this analysis, we have processed a control mouse paw section devoid of CIA immunization treatment. As illustrated in Supplemental Figure S4, the control mouse section deposited on top of a 32 × 64 (2048 printed spot) DNA array displayed a thinner shape than the CIA specimen, in agreement with the consequences of the inflammation effect induced by the immunization. The control paw section has been immunostained with an anti-H3K27ac antibody and exposed to DAPI for nuclei cell density assessment (Supplemental Fig. S4A). By following the aforementioned double-barcoded spatial epigenomic strategy, an Illumina DNA library was obtained and sequenced for a total of more than 450 million reads (Supplemental Fig. S4C,D). The primary bioinformatics analysis obtained more than 120 million reads mapped to the mouse genome, which were used for revealing a spatial digital map by applying a quantile normalization procedure and tissue pattern density processing (Supplemental Fig. S4B–E). In addition to the detection of cartilage progenitor markers in this control sample (Supplemental Fig. S4F), its direct comparison with the CIA-immunized specimen revealed 2599 promoters specific to the control section, 3290 common H3K27ac-enriched promoters, and twice as much promoter regions specifically enriched in the CIA sample (6874 gene promoters) (Supplemental Fig. S4G). Furthermore, a tissue–cell type gene marker association analysis applied to each of these populations confirmed a specific enrichment for the osteoarthritic cartilage tissue (mesenchymal progenitors), or the presence of macrophage synovium signatures in the CIA-specific promoters (Supplemental Fig. S4H).
Among the various CIA H3K27ac overenriched promoters, genes coding for proteins like the transmembrane glycoprotein podoplanin (Pdpn), known to participate in osteocyte function but also an important sensor for bone damage during inflammatory arthritis (Wehmeyer et al. 2019), or the fibroblast growth factor 23 (Fgf23), mainly produced by osteocytes and in elevated levels in rheumatoid arthritis (Hu et al. 2024), were observed across the mouse paw sections (Fig. 3J). Similarly, promoter regions associated to genes like distal-less homeobox 5 (Dlx5), a well-known transcription factor driving osteoblastogenesis (Samee et al. 2008), or gamma-aminobutyric acid type B receptor subunit 1 (Gabbr1), a member of the GABA receptors and known to be overexpressed in rheumatoid arthritis (Song et al. 2024), were also retrieved enriched in H3K27ac signatures across the tissue.
Furthermore, promoter regions associated to genes coding for proinflammatory cytokines, including Il1b, Il6, or Il10, known to drive inflammation and the destructive process of the tissue (Magyari et al. 2014); the cytokines Cd44, Cd166, Cd248 and the colony stimulating factor 1 (macrophage) (Csf1), driving the development of the monocyte–macrophage cell lineage, which play an important function in the pathogenesis of rheumatoid arthritis; or the integrin alpha 2 (Itga2), shown to be overexpressed in RA and influencing T cell growth accompanied by an upregulation of proinflammatory cytokine expression (He et al. 2021) were also enriched for H3K27ac across the studied CIA mouse paws (Fig. 3J). To further validate the H3K27ac enrichment in the aforementioned promoters, a pseudobulk profile was generated from the H3K27ac CIA map and displayed along with public data concerning ChIP-sequencing profiles for the H3K27ac and also RNA sequencing tracks (Supplemental Fig. S5). These public data were obtained from the platform QC Genomics (Mendoza-Parra et al. 2013, 2016; Blum et al. 2020). Furthermore, the pseudobulk profile generated from the H3K27ac CIA maps was stratified into six heatmaps corresponding to the various clusters displayed in Figure 3G, such that the contribution of the various H3K27ac displayed peaks to each of the indicated clusters can be observed (Supplemental Fig. S5).
Finally, to further confirm the relevance of the obtained H3K27ac overenrichment signatures in the CIA specimen, we have compared them with public temporal transcriptomics data obtained from a CIA immunization assay and performed in mice paw soft tissue material (Fig. 3K; Li et al. 2022). The reanalysis of their temporal transcriptomes revealed the presence of seven major gene coexpression paths reflecting the temporal gene expression transitions (Fig. 3L). Importantly, the comparison of these gene coexpression paths with the H3K27ac promoter enrichment signatures retrieved in the CIA specimen revealed up to 50% of common genes with the strongly expressed paths, as well as only ∼10% in the case of coexpression path 6 presenting a downregulated gene expression behavior, confirming a relatively strong correlation between H3K27ac enrichment at the promoter regions and gene expression response (Fig. 3m).
Overall, the aforementioned data clearly demonstrate the capacity of the double-barcoded spatial epigenomic strategy to retrieve RA-related signatures from acid-decalcified FFPE mouse paw samples.
Spatially resolving chromatin-state signatures by integrating multiple histone modification maps generated from consecutive mouse embryo sections
To further illustrate the performance of the presented technology, we have performed spatial epigenomic profiling over mouse embryo sections (E11.5). We have manufactured DNA arrays composed of 32 × 32 × 2 unique double-barcoded probes (32 × 32 interstitially printed arrays, 2048 spots). Mouse embryo sections deposited on top of the DNA arrays were immunolabeled for the histone modification H3K27ac, known to be enriched in active promoters as well as in active enhancer regions. DAPI staining has been used for revealing cell density variability across the tissue section, which has been integrated within the read count normalization (Fig. 4A). A t-SNE analysis revealed six clusters presenting distinct anatomical locations when projecting this information across the spatial SpExel locations (Fig. 4B). Tissue-associated Gene Ontology enrichment analysis confirmed the anatomical classification, illustrated by the spinal cord signature retrieved in clusters 2 and 5; the epicardial progenitor cell hearth association in clusters 1 and 4; a strong nervous tissue association in cluster 5; the presence of olfactory ensheathing cell signatures in cluster 3, known to be derived from neural crest cells and detectable by the expression of Sox10 at E11.5 (Perera et al. 2020); or a ventral otocyst brain signature in clusters 6 and 4; corresponding to progenitor cells of the inner ear detectable at E10.5 by virtue of the expression of the transcription factor Pax2 (Supplemental Figs. S6, S7; Fig. 4C; Durruthy-Durruthy et al. 2014).
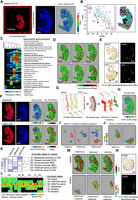
Inferring chromatin-state transitions across the mouse embryo by integrating consecutive spatial epigenomic landscapes. (A, left) Scan of a DNA array (TRITC filter) hosting a cryosection of a mouse embryo (E11.5) and immunostained with an antibody targeting the histone modification H3K27-acetylation (H3K27ac). Notice the presence of Cyanin-3 (Cy3)-labeled DNA probes delimiting the DNA array composed of 32 × 32 interstitially printed probes. (Middle) DAPI staining. (Right) Digital map displaying the normalized read counts captured per physical position (SpExel) across the mouse embryo section. (B) t-SNE analysis allowing to stratify SpExel in six different clusters. (Right) Projection of the stratified clusters within the digital map. (C) Tissue–cell type gene marker association analysis performed per clusters identified in B. (D) Local enrichment signatures associated with six gene promoters presenting H3K27ac overrepresented counts. (E) In situ hybridization (ISH) and gene expression data (Allen Mouse Brain Atlas) for the genes Sox10 and Lhx5, revealing their spatial signature, which is coherent with the H3K27ac digitized view displayed in D. (F, left to right) Histone modification immunostaining (TRITC filter: H3K4me3, H3K27me3), nuclei revealed by DAPI, digital map of the normalized read counts across the tissue section, and local read count enrichment associated to the gene Wnt11. (G) Strategy for interrogating changes in chromatin histone modification signatures across the embryo: (i) overlay all three histone modification maps, (ii) generate a pseudomap in which SpExel of each digitized map are allocated to common pseudocoordinates, and (iii) interrogate for contiguous enrichment patterns similarity within tissue section but also across all three aligned sections. (H) Pseudomap obtained from the overlay of all three histone modification digital maps. (I) Promoter enrichment patterns associated to Wnt11 gene (more than five contiguous SpExel) retrieved in either of the histone modification maps. (J) Spatial gene promoter's coenrichment analysis for Wnt11 in the H3K27ac or H3K27me3 digital map. SpExel colored in red correspond to the location of Wnt11, whereas others correspond to other gene promoters sharing a similar spatial pattern (Tanimoto similarity index). Notice that these two spatial gene promoter's coenrichment maps present distinct spatial localizations. (K) Heatmap displaying the co-occurring promoter enrichment patterns between all three histone modifications. Six chromatin co-occurring states were identified and functionally associated to either active, repressed, or bivalent promoters, as described previously (Ernst and Kellis 2010, 2012). Notice the presence of two states for which no functional association has been attributed: H3K4me3, or H3K27ac alone. (L) Gene promoters presenting different chromatin co-occurring states across the tissue. (M, top) Local read counts promoter enrichment for the gene Hoxb4 in either of the histone modification maps within the pseudomap. (Bottom) Hoxb4 coenrichment patterns revealing the presence of either bivalent (H3K27me3/H3K27ac or H3K27me3/H3K4me3) or promoter active regions (H3K27ac/H3K4me3). (N) In situ hybridization (ISH) and gene expression data (Allen Mouse Brain Atlas) for the gene Hoxb4, revealing its spatial signature coherent with the H3K27ac digitized view displayed in M.
In addition to the H3K27ac local enrichment in the promoter regions of Sox10 or Pax2, other genes like LIM Homeobox 5 (Lhx5), known to be broadly expressed in the early embryo but later restricted to the nervous system (Peng and Westerfield 2006); the transmembrane protein 80 (Tmem80), previously shown to be expressed in the head surface ectoderm (RNA-seq assays in embryonic mice (E8.5, E9.5, E10.5) (Brunskill et al. 2014)); or two members of the Wnt family, Wnt4, previously shown to be expressed in the spinal cord of E9.5 and E10.5 mouse embryos (Agalliu et al. 2009), or Wnt11, shown to be required for proper patterning of the neural tube and somites by regulating notochord formation (Andre et al. 2015) were also enriched for this histone modification (Fig. 4D). The relevance of finding H3K27ac enrichment in the aforementioned gene promoters was also confirmed in the spatial epigenomic cartography previously described (Supplemental Fig. S6; Deng et al. 2022) but also by gene expression data assessed in mouse embryo (E11.5) by the Allen Developing Mouse Brain Atlas (Fig. 4E; Supplemental Fig. S6).
Although several of the retrieved signatures in the spatial H3K27ac cartography do match with previously described gene expression patterns, this histone modification alone is not a fateful marker of active transcription. Indeed, previous studies demonstrated a correlative relationship between the presence of co-occurrent histone modifications at promoter regions with the transcriptional status of the associated gene (Ernst and Kellis 2010, 2012). For instance, promoter regions hosting both H3K4me3 and H3K27ac signatures are known as markers for active transcription, whereas promoters enriched on H3K27me3 correspond to repressed regions. Furthermore, promoters hosting the H3K27me3 modification and either H3K4me3 or H3K27ac are described as “bivalent promoters,” corresponding to genes devoid of transcription activity but prone to initiate gene expression (Ernst and Kellis 2010, 2012). Herein, we aimed to evaluate this potential combinatorial coenrichment signatures (previously described as chromatin-state analysis) (Ernst and Kellis 2010, 2012) for three major histone modifications: H3K4me3, H3K27ac, and H3K27me3. Hence, in addition to the aforementioned H3K27ac spatial cartography, we have also generated spatial epigenomic maps for H3K4me3 and H3K27me3 generated from consecutive sections (Fig. 4F). Because mouse embryo sections are not located in exactly the same physical position on the manufactured DNA arrays, we have computationally realigned all three spatial epigenomic maps, leading to the generation of a spatial pseudomap, used in a second time for interrogating histone enrichment signatures per section but also across all three overlayed sections (Fig. 4G,H). The obtained pseudomap has been first used to verify the spatial coenrichment of the studied histone modifications in the case of the gene Wnt11. Although all three histone modifications were retrieved overenriched in Wnt11 promoter regions (Fig. 4D,F), they do not systematically overlay in their spatial localizations within the embryo, as revealed when interrogating for contiguous overenriched SpExel within the pseudomap. Indeed, distinct spatial localizations within the embryo appeared to be enriched for both H3K4me3 and H3K27ac or the repressive mark H3K27me3 (Fig. 4I). This is further confirmed when interrogating for Wnt11 coenrichment promoter signatures (i.e., other genes presenting the same histone modification enrichments in their promoters and spatially colocalized with Wnt11). Wnt11 H3K27ac/H3K4me3 (active promoter signature) coenrichment patterns were associated to the trunk notochord region, whereas the repressive mark H3K27me3 appears to be enriched to the tail of the embryo (Fig. 4J), in agreement with Wnt11 expression described in previous studies (Supplemental Fig. S7; Andre et al. 2015; Murphy et al. 2022).
Expanding the spatial coenrichment analysis over all genes captured in the spatial epigenomic maps revealed more than 600 promoters presenting H3K4me3/H3K27ac coenrichment signatures (state 3: active promoters), 4759 repressed promoters (state 1: enriched for H3K27me3), and more than 1600 bivalent promoters, defined by their enrichment in H3K27me3 and either H3K4me3 (state 4: 681 genes) or H3K27ac (state 2: 974 genes) (Fig. 4K). Finally, a large fraction of gene promoters were retrieved spatially associated only to H3K4me3 (state 5: 3500) or H3K27ac (state 6: 5140), potentially indicating that we failed to properly associate them to a coenrichment signature owing to technical issues (e.g., more than five contiguous overenriched SpExel, sequencing depth, antibody performance, etc.).
As illustrated for the case of the Wnt11 promoter (Fig. 4I,J), chromatin states in promoter regions do change across tissue section. To characterize these changes, we have selected genes based on the number of overenriched SpExel across the tissue (>40% occupied SpExel when considering all chromatin-state combinations and >10% for at least one of the chromatin states). A total of 750 genes were selected, revealing 291 promoters fluctuating between the active to the repressive state, passing by the bivalent situations. Furthermore, the remaining 459 genes were devoid of the active chromatin-state combination (state 3) but presented the repressive mark H3K27me3 (Fig. 4L). In some cases, these promoters shifted to the bivalent states (62 genes) or replaced the repressive mark by either the “H3K4me3 alone” or “H3K27ac alone” state (80 genes). Furthermore, in some cases, they fluctuated between one of the bivalent states and the presence of either the “H3K4me3 alone” or “H3K27ac alone” state (143 genes hosting the chromatin state S2, 102 genes hosting the S4). Finally, there are 72 genes that do not present the H3K4me3 alone situation but fluctuate among the repressive state (H3K27me3), the bivalent state S2 (H3K27me3/H3K27ac), or the presence of only the histone modification H3K27ac in their promoter regions (Fig. 4L). The fact that these 459 gene promoters are devoid of the active chromatin state but at the same time present either H3K4me3 or H3K27ac alone supports the possibility of missing such a signature owing to technical limitations. For instance, in the case of the Hoxb4 promoter, our spatial epigenomic profiling clearly revealed a strong repressive signature (H3K27me3) other than the spinal cord region. On the contrary, H3K4me3 and H3K27ac profiling do present a spinal cord enrichment localization (Fig. 4m). Hoxb4 gene coenrichment similarity maps revealed a bivalent chromatin-state configuration at the tail of the embryo and a coenrichment signature of the histone modifications H3K4me3 and H3K27ac at the medial level of the spinal cord (Fig. 4m), in agreement with gene expression revealed by in situ hybridization (Allen Developing Mouse Brain Atlas) (Fig. 4n).
Overall, these complex combinatorial chromatin-state transitions clearly reflect tissue complexity but also provides a means to deeply interrogate histone modification signature transitions across the developing system. It is clear that having multiple histone modification profiles and consecutive sections appears essential for further exploiting this molecular cartography.
Discussion
Although recent developments in spatially resolved omics are revolutionizing the way in which we can explore tissue complexity, most of the available solutions are only focused in capturing the presence of RNA. Previous studies indicated that the half-life of RNA molecules within the cell can range from 40 min to 9 h, notably based on its physiological function (Tani et al. 2012), suggesting that a transcriptomic profiling assay does not directly reflect the transcribing status of the cell at the time of its measurement. Furthermore, per definition, a transcriptomic analysis can only identify readouts of expressed genes, but it cannot distinguish between genes being repressed or kept in a prone transcriptional state, because both are transcriptionally inactive.
In bulk assays, the use of chromatin immunoprecipitation strategies for interrogating the presence of histone modifications across the genome became the optimal way to interrogate the transcription regulatory status at the time “t” at which samples are collected (Bannister and Kouzarides 2011). Similarly, the recent development of the enzymatic strategy “Cut&Tag” (based on the use of a recombinant Protein A and the Transposase Tn5) (Kaya-Okur et al. 2019) allowed a gain in sensibility by reducing the number of required cells from millions to few thousands, even reaching the single-cell level (Bartosovic et al. 2021).
In this study, we present a new method that allows the capture of histone modification signatures in a spatially resolved manner by using a double-barcoded DNA array able to capture the PA-Tn5. In this manner, cleaved chromatin is immobilized by creating a physical tagmentation with the DNA probes of the DNA array. Although our technology is not as resolutive as the methodology presented by Spatial-Cut&Tag (20 microns microfluidic channels) (Deng et al. 2022), the flexibility for manufacturing DNA arrays covering custom-based surfaces at a cost-effective manner—owing to the combinatorial number of required probes—makes our approach extremely powerful when aiming to interrogate large number of tissue sections, even when presenting large dimensions (Supplemental Fig. S8). For instance, major efforts, like the one performed by Ortiz et al. (2020) for establishing a molecular atlas of the adult mouse brain by SrT assays in 75 sections (Ortiz et al. 2020), could be expanded to the level of the histone modification signature transitions regulating gene transcription.
Indeed, our technology can be considered as a low-resolution high-throughput screening strategy (i.e., retrieve novel targets through the unbiased power provided by Illumina sequencing) that can be combined with more resolutive methods that require the identification of a potential list of probe panels for interrogating the tissue of interest by in situ hybridization (e.g., Epigenomic MERFISH) (Lu et al. 2022). It is also worth noting that denoising strategies, initially used for image treatment but now getting applied to spatial omics approaches (Jiao et al. 2024), can be combined with our approach to obtain high-resolutive outcomes in a cost-effective manner.
One main limitation of our current technology is that, despite the use of large sequencing-read coverage, a rather small fraction of reads did contribute to the spatial profile, thus leading to rather sparse view across the analyzed sections. By performing multiple assays (fresh-frozen tissues, but also FFPE samples), we have observed that the primary bioinformatic treatment, focused on detecting the dual barcode sequences, represents the current bottleneck of the assay. Hence, instead of investing resources for providing further sequencing depth, we anticipate the elaboration of an enhanced version of our DNA arrays, in which the length of the dual barcodes will be expanded to improve the recovery during the primary steps of the bioinformatic pipeline.
Finally, in this study, we have also highlighted the necessity of collecting consecutive sections for acquiring distinct histone modification signatures, which can then be interrogated across sections for revealing chromatin-state signatures in a spatially resolved manner. We speculate that this strategy will further gain in power in the following years with a decrease in sequencing costs, thus allowing to collect multiple consecutive sections for reconstructing volumetric views of the chromatin-state signatures driving tissue architecture.
Methods
DNA array manufacturing
DNA arrays for spatial epigenomic experiments were produced by depositing two types of complementary oligonucleotides, namely, “barcode for rows” (BCr) and “barcode for columns” (BCc) hosting sequences. Like in our previous study describing the use of double-barcoded DNA arrays for spatial transcriptomics (Lozachmeur et al. 2023), we have used BCr oligonucleotides presenting an amino C6 linker at the 5′ extremity, followed by four G or C nucleotides (S), a T7 promoter sequence (GACTCGTAATACGACTCACTATAGGG), a unique molecular identifier (UMI; WSNNWSNNV), a molecular barcode (8 nt) associated to the printed row in the DNA array, and a 30 nt adapter sequence with a GC-content of 40% (here named as Gibson sequence “ACATTGAAGAACCTG-TAGATAACTCGCTGT”).
The BCc oligonucleotide has been modified for spatial epigenomic applications as follows: At its 5′ extremity, it presents a “CTGTCTCTTATACACATCT” (herein called as a mosaic sequence [MOS]), corresponding to the sequence recognized by the transposase Tn5 (Reznikoff 2003). Then similarly to previous study, BCc contains a unique molecular identifier (UMI WSNNWSNNV), a molecular barcode (8 nt) associated to the printed column in the DNA array, and a complementary Gibson sequence.
Oligonucleotides were diluted to 5 µM in presence of sciSPOT Oligos B1 solution (Scienion CBD-5421-50) in a 96 well plate and stored at −20°C for long storage. DNA arrays were printed onto Superfrost plus adhesion microscope slides (Epredia J7800AMNZ).
For mouse embryo assays, we have manufactured DNA arrays composed by 32 × 32 × 2 interstitially printed probes (2048 different probes in total, 177 µm pitch distance, ∼100 µm printed spot). For this, 32 BCr oligonucleotides presenting a unique molecular barcode were printed per row, by depositing ∼250 pL (250 µm between contiguous spots) with a DNA picoliter spotter (Scienion sciFLEXARRAYER S3). Similarly, 32 different BCc oligonucleotides were printed per column, by spotting ∼250 pL of each of them on top of the previously printed BCr nucleotides. Then, the same 32 BCr oligonucleotides were printed per row with a shifted position of 125 µm in both axis (i.e., interstitial printing), followed by the deposition of 32 different BCc oligonucleotides printed per column. In that manner, a total 32 unique BCr (printed twice) and unique BCc oligonucleotides are required for reaching an array presenting 2048 unique double-barcoded probes.
For mouse brain sections, we have generated 64 × 32 × 2 interstitially printed arrays (4096 different probes), in which 32 BCr oligonucleotides presenting a unique molecular barcode were printed per row; another 32 different BCr oligonucleotides were printed per row with a shifted position of 125 µm in both axis (i.e., interstitial printing), followed by the deposition of 64 different BCc oligonucleotides printed per column. Finally, for mouse paw studies, a simpler DNA array printout was produced (64 × 32 DNA arrays) by depositing 32 BCr probes per row and 64 BCc probes per column (250 microns pitch distance, 100 microns spot diameter).
In all printout designs, fiducial borders were printed per DNA array by adding three row/columns of spots with the used pitch distance (250 µm) and printing a BCr1 oligonucleotide presenting a Cy3-label at the 3′-extremity. During fiducial manufacturing, some spots were skipped on purpose for creating asymmetry, which will help for image scanning.
UV irradiation (254 nm; 5 min) was applied for cross-linking, followed by T4 DNA polymerase elongation (depending on the size of the printing area: 15 µL to 40 µL per printed region with coverslips: 0.06 U/µL T4 DNA polymerase [New England Biolabs M0203L]; 0.2 mM dNTPs [Life Technologies R0141, R0151, R016, R0171] for 1 h at 37°C). To evaluate the double-strand DNA elongation performance, a quality-control assay was made within every batch production (18 slides per batch, two printed regions per glass slide for size areas of 8 × 16 mm [32 × 64 either linear or interstitially printed spots] or three printed areas for 8 × 8 mm arrays [2048 spots]) by incorporating a dCTP-Cy3 nucleotide during the elongation (dATP/GTP/TTP at 0.2 mM, dCTP at 0.005 mM, and dCTP-Cy3 at 0.002 mM; Cytiva PA53021) and analyzed by fluorescent microscopy. After elongation, slides were washed with 0.1 × SSC buffer (Merck S6639) and then ddH2O and finally spin dried (Labnet slide spinner C1303-T-230V, 4800 rpm). DNA array slides have been stored at 4°C in a sealed container.
Fresh-frozen tissue cryosectioning
Tissue samples were cryosectionned (16 µm sections for mouse embryo tissue and and 20 µm sections for mouse brain WT tissue), collected between −15°C and −12°C, and then deposited on the DNA-arrays defined regions. Tissue sections were fixed with 1% para-formaldehyde (PFA; Life Technologies 28908) in 1 × PBS (Life Technologies 70011-036) for 30 min at room temperature and then washed three times with 1 × PBS.
Mouse paw decalcification, formalin-fixed embedding, and sectioning
Mouse paws were collected and fixed in 4% buffered formol in 1 × PBS for 24 h. Then, paws were decalcified for seven consecutive days in an 4°C refrigerated room on agitation. A maximum of five paws, each inside a cassette, were decalcified in 1 L solution 3% formic acid in PBS. At the mid incubation time, the decalcification solution was renewed and paws were cut in half and put back into the cassette. Once decalcification was finished, the paws were rinsed in ddH20 to be dehydrated and infiltrated with paraffin using an automated device (ASP300 Leica microsystems) following the following standard protocol: 70% ethanol, two washes of 70% ethanol, 96% ethanol, 100% ethanol, half and half mix of 100% ethanol with 100% xylene, empty the incubation chamber, a final step of 100% xylene, and three baths of paraffin to allow paraffin infiltration. Afterward, each half paw has been embedded in paraffin blocks. All blocks of mouse paw FFPE can be stored at room temperature.
The 4 µM thick sections around the talus bone were collected from those FFPE mouse paw blocks using a microtome. Paw FFPE sections were deparaffined and rehydrated following standard protocols on an automated device (BondRx, Leica Microsystems).
Histone modifications associated with chromatin capture in situ
Tissue sections were permeabilized for 1 h at room temperature with 1% Triton X-100 (Merck 93443) in 1 × PBS solution, washed three times with 1 × PBS followed by a 5 min incubation at room temperature with 0.1 m HCl for chromatin loosening as described previously (Lu et al. 2022), and washed three times with 1 × PBS.
Afterward, sections were covered for 1 h at room temperature with a blocking and permeabilizing mix (10% normal donkey serum [NDS; Merck D9663], 0.25% Triton X-100 [Merck 93443], 0.1% bovine serum albumin [BSA; Merck A9418], 2 mM EDTA [Life Technologies AM9260G], 0.5 mM spermidine [Life Technologies 132740010], one tablet EDTA-free protease inhibitor cocktail [PIC; Merck 11873580001]) prepared in 1 × PBS. Then, tissues were washed twice with 150 mM NaCl buffer (20 mM HEPES-KOH 1 m at pH 7.2 [Merck 391338], 150 mM NaCl; 0.1% BSA, 0.5 mM spermidine, one tablet PIC).
After that, sections were incubated with a solution containing 2 μg of a selected primary antibody (antihistone H3 [trimethyl K4] antibody [Abcam ab213244]; antihistone H3 [acethyl K27] antibody [Abcam ab177178]; or antihistone H3 [trimethyl K27] antibody [Abcam ab 192985]), in 150 mM NaCl buffer for 1 h at room temperature followed by washing twice with 150 mM NaCl buffer. Then, tissues were incubated at room temperature with a preincubated mix (2 h) containing 1:1 molar ratio of unloaded fusion protein pA-Tn5 (in-house production [9.5 µM] prepared using the plasmid Addgene 124601) by following previous protocols (Kaya-Okur et al. 2019; Li et al. 2021) and the corresponding secondary antibody (donkey antirabbit IgG antibody; Merck SAB3700932). The preincubated mix was complemented with either a conjugated Alexa Fluor 488 secondary antibody (donkey antirabbit IgG antibody, Alexa Fluor 488 [Life Technologies A-21206]) or a conjugated Alexa Fluor 555 secondary antibody (donkey antirabbit IgG antibody, Alexa Fluor 555; Abcam ab 150062), added at 1/1000 dilution.
The preincubated mix was used in a five times molar excess relative to the number of moles of the MOS sequence deposited per DNA array: For 2048 spots, it corresponds to 2.56 × 10−12 moles; for 4096 spots, it corresponds to 5.12 × 10−12 moles.
After 3 h of incubation with the assembled mix, tissue sections were washed twice with 300 mM NaCl buffer (20 mM Hepes-KOH 1 m at pH 7.2, 300 mM NaCl; 0.1% BSA, 0.5 mM spermidine, one tablet PIC) to remove PA-Tn5 antibodies in excess. Then, tissue sections were incubated for 5 min with DAPI (Life Technologies 62248) at 1/5000 dilution in 1 × PBS and then washed twice with 1 × PBS.
DNA arrays were scanned under the TRICT filter to reveal the presence of the fiducial borders as well as the physical position of tissue sections on top of the printed DNA arrays. In case of the use of the conjugated Alexa Fluor 488 secondary antibody, an additional scanning using FITC filter was done to reveal the localization of the studied modification of histone. Finally, an additional scanning of the tissue using DAPI filter was done to observe nuclear localization and to get cell density information. After imaging, sections were incubated overnight at 37°C with 10 mM MgCl2 (Life Technologies AM9530G) in 300 mM NaCl buffer for in situ tagmentation.
The next day, tissue sections were incubated with a mix containing 2.25 µL of EDTA 0.5 m, 2.75 µL of 10% sodium dodecyl sulfate (SDS; Merck 71736), and 0.5 µL of 20 mg/mL Proteinase K (Life Technologies 25530049) in 50 µL of ddH2O overnight at 37°C. The mix volume was adjusted depending on the slide printing area.
Finally, slides were washed under agitation (300 rpm) in containers containing 100 mL of preheated buffer 1 (2 × SSC and 0.1% SDS) for 15 min at 50°C and then 10 min with buffer 2 (0.2 × SSC) at room temperature and 10 min with buffer 3 (0.1 × SSC) at room temperature. Lastly, the slides were washed in ddH2O and spin-dried.
After that, DNA arrays were incubated with 0.1 m NaOH solution for 10 min at room temperature and then neutralized after removing liquid with a mix containing 100 µL of 0.1 m NaOH, 11.8 µL of 10 × TE, and 6.5 µL of 1.25 m acetic acid for 2–3 min. Finally, slides were washed within a Falcon containing buffer 3 (0.1 × SSC) and then into a Falcon containing ddH2O and spin-dried.
cDNA complementary strand synthesis
DNA arrays were incubated with a poly(C) tailing mix containing 0.6 U/µL terminal transferase (New England Biolabs M0315), 1 × terminal transferase buffer (New England Biolabs BO315), 0.3 × CoCl2 (New England Biolabs B0252), 0.2 mM dCTP, and 0.01 mM ddCTP for 35 min at 37°C and then 20 min at 70°C and cooled at 12°C. To avoid evaporation, each printing area was closed by using sealing chambers (size 25 μL, Life Technologies AB-0576; size 65 μL, Life Technologies AB-0577). Finally, slides were washed within a Falcon containing buffer 3 and a Falcon containing ddH2O, before spin-drying.
DNA arrays were then incubated with a mix containing 0.1 U/µL Klenow exo- (New England Biolabs M0212), 0.2 mg/mL BSA, 1 × NEB2 buffer (New England Biolabs B7002), 0.5 mM dNTPs, and an oligonucleotide (1 µM) having a complementary sequence for the poly(C) tailed sequence, as well as a 5′-extremity providing an adapter sequence (“GTTCAGACGTGTGCTCTTCCGATCTGGGGGGGGGH”). As in the preceding step to avoid evaporation, the reaction was performed by using sealing chambers. The following conditions were used: 5 min at 47°C (primer annealing), 1 h at 37°C (extension), 10 min at 70°C (enzyme deactivation), and cooling at 12°C. To recover the synthesized complementary DNA strands, DNA arrays were incubated with 100 µL of 0.1 m NaOH solution for 10 min, and then the liquid was collected and a second incubation with 0.1 m NaOH solution was repeated. The two fractions were collected and neutralized (for 100 µL 0.1 m NaOH solution, add 11.8 µL of 10 × TE and 6.5 µL of 1.25 m acetic acid). DNA arrays were also neutralized with 100 µL 0.1 m NaOH solution mixed with 11.8 µL of 10 × TE and 6.5 µL of 1.25 m acetic acid and then washed with buffer 3 and ddH2O and stored at 4°C for eventual supplemental experiments.
The neutralized collected solution was precipitated with cold 100% ethanol (2 × volume), 200 mM NaCl, and 1 µL of 20 mg/mL glycogen (Merck 10901393001); conserved at −20°C for at least 1 h; centrifuged at 12,500 rpm for 30 min; washed once with cold 70% ethanol; and resuspended (after 30 min air drying) in 23 µL of ddH2O.
Illumina sequencing library preparation
Half of the resuspended cDNA's complementary-probe material (12 µL) was adjusted to 23 µL with water and then mixed with 25 µL of Q5 hot start high fidelity 2 × master mix (New England Biolabs M0464L), 1 µL of adapter seq1 primer (0.02 µM; GTTCAGACGTGTGCTCTTCCGATCT), and 1 µL of adapter seq2 primer (0.02 µM; TACACTCTTTCCCTACACGACGCTCTTCCGATCTGACTCGTAATAC, corresponding to a part of the T7-promoter sequence). This mix was amplified by PCR (30 sec at 98°C; 15 cycles of 10 sec at 98°C and 75 sec at 65°C; 5 min at 65°C; 12°C hold), cleaned at 0.9 × ratio with SPRIselect beads (Beckman Coulter B23318), and resuspended in 24 µL of ddH2O.
A second PCR amplification was done by mixing the 24 µL of adaptors linked cDNA's complementary-probe strand with 25 µL of Q5 hot start high fidelity 2 × master mix, 0.5 µL of universal primer, and 0.5 µL of index primer (0.1 µM final concentration NEBNext multiplex oligos for Illumina index primers set 1 E7335) by using the following cycling conditions: 30 sec at 98°C, 15 cycles of 10 sec at 98°C and 75 sec at 65°C, a final extension at 65°C for 5 min, and a hold at 12°C.
Then sample was cleaned at 0.9 × ratio with SPRIselect beads. Finally, the library was used for Illumina sequencing (150 nt paired-ends sequencing; NovaSeq 6000; final loading concentration, 70 pM; pool loading concentration, 0.35 nM; XP-mode).
Bioinformatics processing
Primary analysis has been performed with our in-house developed tool SysFate Illumina Spatial transcriptomics Demultiplexer (SysISTD) available at GitHub (https://github.com/SysFate/SysISTD) and as Supplemental Code.
SysISTD takes as entry paired-end sequenced reads (FASTQ or FASTQ.gz format), and two TSV files, the first one containing the sequence of the molecular barcodes associated to the rows or column in the printed arrays and the second file presenting the physical position architecture of the spatial barcodes. SysISTD searches for the Gibson sequence (regex query) and then for two neighboring barcodes. Paired reads presenting these features were aligned to the mouse (mm9) genome with Bowtie 2 (Langmead and Salzberg 2012). In this manner, SysISTD generates SAM files per spatial coordinate, which are reprocessed with the help of featureCounts (Liao et al. 2014) and an in-house script for detecting reads counts per gene promoter (±5 kb) and per spatial coordinate SysFate Illumina Spatial Promoters Demultiplexer (SysISPD; https://github.com/SysFate/SysISPD). Although the mouse reference genome mm10 has been available since December 2011, we are still working with the mm9 version because we systematically perform a direct comparison of our findings with the NGS-QC database, composed by more than 100,000 publicly available bulk ChIP-sequencing data built on the mouse mm9 / human hg19 reference genomes. Furthermore, >90% of the genes retrieved in the mm10 version are already available in the mm9 genome assembly.
As an outcome, SysISPD generates a matrix presenting read counts associated to physical coordinates in columns and known transcripts ID associated to the evaluated promoters in rows. To focus the downstream analysis to the physical positions corresponding to the analyzed tissue, we used an in-house R script taking as entry an image of the DNA array scanned with the TRICT filter, revealing the presence of the fiducial borders.
Specifically, we uploaded to R (R Core Team 2020) a cropped image within the fiducials (imager package), and we used the “px.flood” function to retrieve the pixels associated to the tissue. Finally, we applied a pixel to gexel coordinates conversion prior to cross this information with the outcome of SysISPD.
In parallel, all SAM files per coordinate were merged into a single-file and processed with the peak caller MACS2 (Zhang et al. 2008). The obtained confident peaks (P-value < 1 × 10−5) were annotated to their proximal coding regions. Finally, the spatial coordinate/promoter matrix provided by SysISPD was filtered by selecting transcripts associated to promoter regions presenting confident peaks.
The “confident promoter peaks tissue-focused” matrix was quantile-normalized with our previously described tool MULTILAYER (Moehlin et al. 2021a,b). Then, the normalized matrix was further processed for revealing cell/tissue density contrast information by integrating normalized counts with pixel value levels retrieved in either the DAPI or the histone modification immunostaining image. The obtained normalized and pixel-intensity-adjusted matrix was used for all downstream analysis.
A t-SNE analysis has been performed within R with the help of the Rtsne package. Projection of the stratified clusters (k-means metrics) was performed with the help of an in-house R script. Genes presenting overrepresented counts per promoter and per cluster were used for performing a cell/tissue type enrichment analysis with Enrichr (Chen et al. 2013) and by using the Mouse Gene Atlas, Tabula Muris, ARCHS4 (Lachmann et al. 2018), CellMarkerAugmented (Zhang et al. 2019), and Panglao (Franzén et al. 2019) databases.
Mouse embryo section alignment and chromatin state transition detection
SpExel physical positions for each mouse embryo section were realigned relative to the tail of the embryo, allowing us to associate the same pseudocoordinates for all the evaluated sections.
The associated histone modification identifier was added to the name of each transcript within the aligned matrices and concatenated into a single matrix, allowing interrogation of contiguous SpExel within sections and enrichment patterns similarity across sections, as described previously in MULTILAYER (Moehlin et al. 2021a,b). Contiguous promoter patterns and promoter coenrichment patterns were captured by MULTILAYER over the embryo pseudomap hosting data of all three histone modifications. Chromatin-state transitions were computed by extracting promoter coenrichment patterns similarity over all three sections with at least five consecutive SpExel.
Data access
All data sets generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE286251. Furthermore, processed spatial matrices as well as the corresponding scanned images are available in the Mendeley Database (https://data.mendeley.com/datasets/d73mg855tw/4).
Competing interest statement
There is a patent application (no. EP24307128) pending for M.A.M.-P. The other authors declare no competing interests.
Acknowledgments
We thank all members of the team SysFate for contributing to the discussion of this project and the Genoscope sequencing platform for their technical support. Notably, we thank Ms. Sandrine Lebled, Isabelle Bordelais, and Alice Moussy from the Genoscope sequencing platform for finding the optimal conditions for the deposition of Illumina libraries within the sequencing instrument. This work was supported by the institutional bodies CEA, CNRS, and Université d'Evry-Val d'Essonne. M.G.M.-F. has been funded by the Institut National du Cancer (INCa: funding 2020-124; 2022-078), G.L. by the ANRT doctoral fellowship CIFRE 2021/1899 in collaboration with Novalix. M.D. is supported by EU grant HORIZON 101070740. Mouse embryos were kindly provided by Wojciech Krezel (IGBMC, Strasbourg, France). Mouse brain samples were kindly provided by Jean-Pierre Mothet (LuMin, ENS Paris Saclay, France). Mouse paws were provided by Novalix.
Author contributions: M.G.M.-F. performed formal analysis, investigation, and methodology. G.L. provided resources and methodology. M.D. performed data curation, software, and formal analysis. L.P. provided resources and methodology. D.M. provided resources and methodology. A.G. performed supervision, review, and editing. M.A.M.-P. performed conceptualization, formal analysis, supervision, funding acquisition, and writing the original draft.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280305.124.
-
Freely available online through the Genome Research Open Access option.
- Received December 2, 2024.
- Accepted April 23, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








