
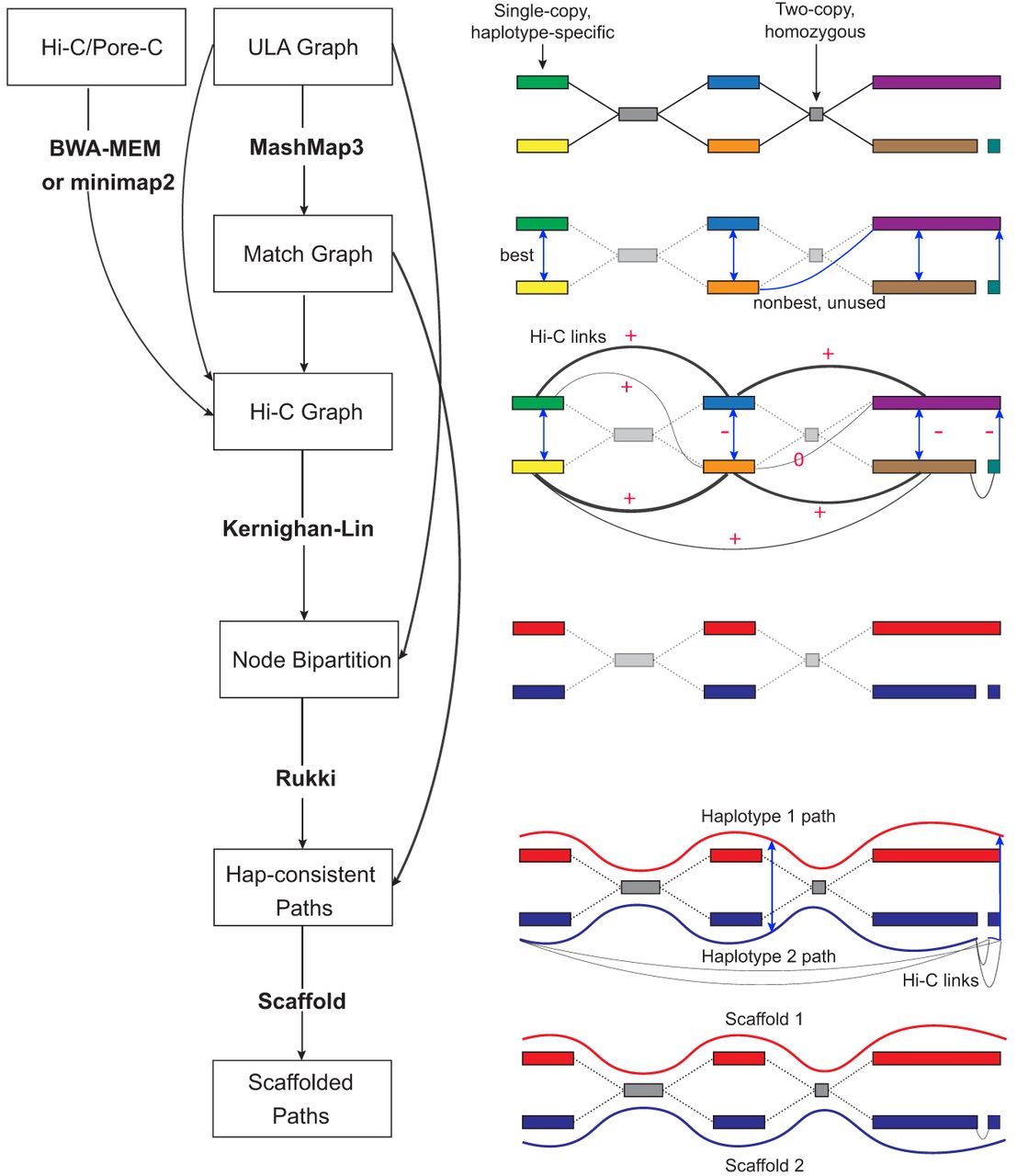
Overview of the Verkko2 Hi-C/Pore-C processing. The process starts with the ULA graph built from the LA and UL sequences. Note that the ULA graph links are only used to cluster the graph into connected components so they are shown as dotted gray lines in the figure. The BWA-MEM/minimap2 step aligns the Hi-C or Pore-C data to the sequences of the ULA graph nodes, and counts connecting pairs of nodes are tallied. Next, the Match Graph step ignores homozygous (based on coverage) and short (by default ≤200 kb) nodes. The remaining nodes are self-aligned to identify homology. The initially computed Hi-C edges are filtered using the Match Graph to build the Hi-C Graph. In many cases, the highest-count Hi-C connection is between homologous pairs of nodes. To avoid these false links, edges connecting potentially repetitive nodes are removed (shown with a value of zero), whereas edges connecting homologous nodes are given large negative weights (shown with −). These updated link weights are used to bipartition each connected component of the graph into two haplotypes. These partitions are then provided to Rukki along with the ULA graph to generate haplotype paths, and the pipeline proceeds as in Verkko1. These haplotype-consistent paths are again used to identify homology, shown with blue edges based on the Match Graph step. All four possible connections are considered to connect the two blue paths based on Hi-C link evidence. In this example, the alignment of both blue paths to the red path adds a multiplicative bonus to one Hi-C connection consistent with the alignment, leading to a scaffold connecting the blue paths.








