
Genetic effects on chromatin accessibility uncover mechanisms of liver gene regulation and quantitative traits
- Kevin W. Currin1,
- Hannah J. Perrin1,
- Gautam K. Pandey1,
- Abdalla A. Alkhawaja1,
- Swarooparani Vadlamudi1,
- Annie E. Musser1,
- Amy S. Etheridge1,2,
- K. Alaine Broadaway1,
- Jonathan D. Rosen1,
- Arushi Varshney3,
- Amarjit S. Chaudhry4,
- Paul J. Gallins5,
- Fred A. Wright5,6,7,
- Yi-hui Zhou5,6,
- Stephen C.J. Parker3,8,9,
- Laura M. Raffield1,
- Erin G. Schuetz4,
- Federico Innocenti2 and
- Karen L. Mohlke1
- 1Department of Genetics, University of North Carolina, Chapel Hill, North Carolina 27599, USA;
- 2Eshelman School of Pharmacy, Division of Pharmacotherapy and Experimental Therapeutics, University of North Carolina, Chapel Hill, North Carolina 27599, USA;
- 3Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 4Department of Pharmaceutical Sciences, St. Jude Children's Research Hospital, Memphis, Tennessee 38105, USA;
- 5Bioinformatics Research Center, North Carolina State University, Raleigh, North Carolina 27695, USA;
- 6Department of Biological Sciences, North Carolina State University, Raleigh, North Carolina 27695, USA;
- 7Department of Statistics, North Carolina State University, Raleigh, North Carolina 27695, USA;
- 8Department of Human Genetics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 9Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, Michigan 48109, USA
Abstract
Chromatin accessibility quantitative trait locus (caQTL) studies have identified regulatory elements that underlie genetic effects on gene expression and metabolic traits. However, caQTL discovery has been limited by small sample sizes. Here, we map caQTLs in liver tissue from 138 human donors and identify caQTLs for 35,361 regulatory elements, including population-specific caQTLs driven by differences in allele frequency across populations. We identify 2126 genetic signals associated with multiple, presumably coordinately regulated elements. Coordinately regulated elements link distal elements to target genes and are more likely to be associated with gene expression compared with single-element caQTLs. We predict driver and response elements at coordinated loci and find that driver elements are enriched for transcription factor binding sites of key liver regulators. We identify colocalized caQTLs at 667 genome-wide association (GWAS) signals for metabolic and liver traits, and annotate these loci with predicted target genes and disrupted transcription factor binding sites. CaQTLs identify threefold more GWAS colocalizations than liver expression QTLs (eQTLs) in a larger sample size, suggesting that caQTLs can detect mechanisms missed by eQTLs. At a GWAS signal colocalized with a caQTL and an eQTL for TENM2, we validated regulatory activity for a variant within a predicted driver element that is coordinately regulated with 39 other elements. At another locus, we validate a predicted enhancer of RALGPS2 using CRISPR interference and demonstrate allelic effects on transcription for a haplotype within this enhancer. These results demonstrate the power of caQTLs to characterize regulatory mechanisms at GWAS loci.
Chromatin accessibility quantitative trait loci (caQTLs) have successfully identified functional variants and regulatory elements at a subset of gene expression QTLs (eQTLs) and genome-wide association study (GWAS) signals (Degner et al. 2012; Kumasaka et al. 2016, 2019; Alasoo et al. 2018; Bryois et al. 2018; Gate et al. 2018; Currin et al. 2021; Liang et al. 2021). However, few caQTL studies have been conducted with samples from 100 or more individuals (Bryois et al. 2018; Gate et al. 2018; Kumasaka et al. 2019), and all of these used brain or blood. Given that GWAS variants are enriched in regulatory elements of trait-relevant tissues (Roadmap Epigenomics Consortium et al. 2015), larger caQTL studies in additional tissues are needed to predict mechanisms at additional GWAS signals. Further, population differences in caQTL discovery have not been well characterized.
Most caQTLs have not been linked to an eQTL (Degner et al. 2012; Currin et al. 2021; Liang et al. 2021), which may reflect the more direct effects of genetic variants on regulatory elements than on gene expression. Consistent with this principle, genetic variants have been shown to explain a larger proportion of variation in chromatin accessibility than gene expression (Liang et al. 2021). Additional approaches, such as Hi-C (Jung et al. 2019) and chromatin coaccessibility (Thurman et al. 2012; Kumasaka et al. 2019; Currin et al. 2021), can be used to predict target genes at caQTLs currently lacking evidence of a shared eQTL.
Previous studies have identified coordinately regulated elements that share the same caQTL signal, and coordination between distal and promoter elements has been used to link distal elements to target genes (Grubert et al. 2015; Waszak et al. 2015; Chen et al. 2016; Kumasaka et al. 2016, 2019; Alasoo et al. 2018; Gate et al. 2018; Delaneau et al. 2019). However, identifying the “driver” elements responsible for these signals remains challenging because caQTL variants may overlap multiple elements at a coordinated signal (Alasoo et al. 2018), and methods for inferring causal direction require sample sizes that are larger than many existing caQTL studies (Kumasaka et al. 2019). Determining if specific transcription factors (TFs) preferentially bind driver elements may further understanding of how these coordinately regulated element sets are formed, and identifying driver elements will help fine-map causal elements and variants at colocalized GWAS loci (van Mierlo et al. 2023).
The liver influences many human traits and diseases through its role in protein synthesis, lipid and glucose metabolism, detoxification, and drug metabolism (Almazroo et al. 2017; Trefts et al. 2017). GWAS loci for metabolic, inflammatory, and other traits are enriched in liver regulatory elements (Roadmap Epigenomics Consortium et al. 2015) and/or colocalized with liver eQTLs, caQTLs, or histone QTLs (Çalışkan et al. 2019; Etheridge et al. 2020; The GTEx Consortium 2020; Currin et al. 2021). We previously identified 3123 caQTLs in liver tissue using only 20 individuals, including 110 caQTLs that colocalized with GWAS signals (Currin et al. 2021). These results suggested that mapping liver caQTLs in additional individuals would substantially increase caQTL discovery, allow characterization of coordinated caQTLs, and facilitate elucidation of mechanisms of additional GWAS signals.
In the current study, we aimed to map caQTLs in an expanded sample size to increase the catalog of genetic effects on liver regulatory elements. We used this expanded set of caQTLs to identify coordinated regulatory elements that share the same genetic signal and to investigate their driver elements. Finally, we integrated caQTLs with external genomic data sets and functional assays to predict and validate mechanisms at GWAS signals.
Results
Genetic effects on liver chromatin accessibility
We profiled chromatin accessibility using ATAC-seq (Buenrostro et al. 2013; Corces et al. 2017) in liver tissue samples from 138 human individuals, consisting of 89 males and 49 females (Fig. 1A; Supplemental Table 1). The ages of donors ranged from 2 to 81 years, and the mean age was 44 years. Among the 109 donors with reported height and weight, the mean body mass index (BMI) was 28 (standard deviation, 7.7). Using genotype principal component analysis (PCA) to infer genetic similarity, we identified 118 individuals similar to European, 17 individuals similar to African, and three individuals similar to admixed populations from The 1000 Genomes Project (Fig. 1A; Supplemental Fig. 1; Supplemental Table 1; The 1000 Genomes Project Consortium 2015). We generated an average of 85 million high-quality aligned ATAC-seq read pairs per individual (Supplemental Tables 2, 3) and identified 358,304 accessible chromatin regions (peaks) present in at least seven individuals (5% of sample size) (Supplemental Data). We replicated 91% of the peaks identified in our previous 20-individual study (Currin et al. 2021), which had similar sequencing depth (about 102 million aligned read pairs/individual) and 93% of liver tissue ATAC peaks mapped in eight individuals from ENCODE (Supplemental Data; The ENCODE Project Consortium et al. 2020); 111,349 (31%) of the peaks in the current study were not present in either prior study, demonstrating the utility of an increased sample size for regulatory element discovery.
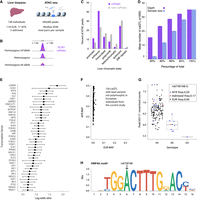
Mapping and characterizing caQTLs in liver tissue. (A) Liver chromatin accessibility data generated by ATAC-seq. (B) caQTL mapping strategy testing variants within 1 kb of ATAC peak centers. (C) caPeaks (FDR < 5%; purple) and non-caPeaks (FastQTL beta-adjusted P > 0.5; gray) overlapping chromatin states from liver tissue from the NIH Epigenomics Roadmap. (D) Number of caQTLs identified after downsampling read depth or sample size to various percentages of full depth. Mean (bars) and standard deviation (error bars) for five technical replicates. Approximate number of read pairs used in each downsampled percentage: 17 million, 34 million, 51 million, and 68 million. Number of samples used in each downsampled percentage: 27, 55, 82, and 110. (E) TF motifs more often disrupted by caQTL variants than non-caQTL variants. Log odds ratios and 95% confidence intervals. Only motifs with P < 1.8 × 10−4 and odds ratio (OR) ≥ 2 are shown. Full results are shown in Supplemental Table 8. (F) Comparison of minor allele frequencies (MAFs) in TOPMed African (AFR) and European (EUR) populations for the lead variants of the 156 caQTLs that are not polymorphic in the individuals genetically similar to European populations. (G) caQTL for caPeak165117 with lead variant rs6758168 not detected in individuals genetically similar to European populations. The G allele, associated with lower chromatin accessibility, was observed in individuals genetically similar to African (blue) and admixed (red), but not European populations. Frequencies correspond to individuals in this study. The boxplots represent ATAC peak counts normalized for covariates. (H) Sequence logo motif for HNF4A, which is disrupted by rs6758168 (position shown by arrow). The G allele is predicted to result in a weaker motif match.
To identify peaks that vary across individuals owing to genotype, we tested for caQTLs using genetic variants within 1 kb of the centers of autosomal peaks and identified 35,361 peaks with a significant caQTL (caPeaks; FDR < 5%) (Fig. 1B; Supplemental Figs. 2, 3; Supplemental Data; Supplemental Table 4). The ATAC PCs used as caQTL covariates were correlated with ATAC technical metrics, including the percentage of reads in peaks for PC1 and the percentage of mitochondrial alignments for PC2, as well as with phenotypes, including age and BMI for PC3 (Supplemental Table 4B). We replicated 2438 caQTLs (78%) from our previous caQTL study (Supplemental Table 5; Currin et al. 2021). Compared to non-caPeaks (FastQTL beta-adjusted P-value > 0.5) (Ongen et al. 2016), a higher percentage of caPeaks overlapped promoter and enhancer chromatin states in the liver, and a lower percentage overlapped other chromatin states (Fig. 1C; Supplemental Table 6).
Although the effect of increased sample size on QTL discovery is well established (The GTEx Consortium 2020), the effect of sequencing depth on QTL discovery is less apparent. Based on sequencing depth downsampling, we identified 2.3-fold more caQTLs in the full-depth data (about 85 million read pairs/individual) relative to the average number of caQTLs at 20% of sequencing depth (n = 15,539) (Fig. 1D; Supplemental Table 7A). Based on sample size downsampling, we identified 17.6-fold more caQTLs using all samples (n = 138) compared with the average number of caQTLs at 20% of sample size (Fig. 1D; Supplemental Table 7B). For sequencing depth, the number of caQTLs gained between 20% and 40% was 2.7-fold higher than the number gained between 80% and the full depth (Fig. 1D; Supplemental Table 7A). For sample size, the number of caQTLs gained between 20% and 40% was only 1.4-fold more than that gained between 80% and the full sample size (Fig. 1D; Supplemental Table 7B), suggesting that we are approaching saturation in sequencing depth more quickly than saturation in sample size. These results suggest that sample size has a larger effect on caQTL discovery than sequencing depth but that increasing sequencing depth is a valuable approach for increasing caQTL discovery when additional samples are not available.
To predict TFs that may regulate chromatin accessibility in liver, we tested for TF binding motifs that are more often disrupted by caQTL variants compared with non-caQTL variants. We identified motifs for 78 TFs whose disruption is significantly associated with caQTL status (odds ratio [OR] > 1, P < 1.8 × 10−4) (Fig. 1E; Supplemental Table 8; Supplemental Data). These include TFs known to alter chromatin accessibility, including FOXA pioneer factors (Mayran and Drouin 2018) and the key liver regulators HNF1A, HNF4A, and ONECUT1 (Nagaki and Moriwaki 2008). These results help predict the most critical TFs for regulating liver chromatin accessibility. Further, variants that disrupt motifs for these TFs may be more likely to be functional, aiding fine-mapping variants at GWAS loci.
We next used disrupted TF motifs to predict how often higher chromatin accessibility was linked to stronger TF binding. Among motif occurrences predicted to be disrupted by caQTL variants (Supplemental Data), 61% of more accessible alleles had a stronger motif match. When using 5368 occurrences of the 40 motifs whose disruption was strongly associated with caQTL status (OR > 2, P < 1.8 × 10−4) (Fig. 1E), 84% of more accessible alleles had a stronger motif match. These results are consistent with previous findings that most TF binding occurs in accessible chromatin (Thurman et al. 2012).
We next examined caQTL differences by population. We identified 156 caQTLs in which the lead variant was not polymorphic (minor allele frequency [MAF] = 0) in individuals similar to European populations (Fig. 1F; Supplemental Data), indicating that these caQTLs were only detected in individuals similar to African populations, which constituted almost all our remaining participants. Consistent with this hypothesis, all of these lead variants are common in TOPMed African populations (MAF range of 12%–50%), and the vast majority (95%) are low frequency (MAF < 1%) in TOPMed European populations. We identified disrupted TF motifs within caPeaks for 64 of these 156 caQTLs, suggesting that population differences in allele frequency may lead to differential TF binding at these loci. At an example in caPeak165117 (Fig. 1G), the G allele of the lead caQTL variant rs6758168 overlaps the caPeak and is associated with lower chromatin accessibility and results in a weaker motif match for HNF4A compared with the A allele (Fig. 1H); the G allele has an allele frequency of 0.29 in the donors similar to African populations in the current study and is not observed in the donors similar to European populations in the current study. Taken together, these results indicate that differences in allele frequency between populations can result in population-specific caQTLs.
Coordinately regulated peaks
Given the relatively large sample size of this study, we sought to identify and characterize caQTL signals associated with multiple regulatory elements. To better identify long-range genetic effects on multiple peaks, we mapped caQTLs using variants within 1 Mb of peak centers and identified 21,074 significant caQTLs (FDR < 5%) (Fig. 2A; Supplemental Fig. 3; Supplemental Table 4); an increased multiple testing burden per peak resulted in fewer caQTLs compared with the 1 kb analysis. After clumping the lead variants (linkage disequilibrium [LD] r2 > 0.9), we identified 2126 signals that are associated with two or more caPeaks (Supplemental Table 9), corresponding to 6077 unique caPeaks. We refer to caPeaks that share a genetic signal with another caPeak as coordinated caPeaks. The largest set contained 47 peaks, and 213 sets contained five or more peaks (Fig. 2B; Supplemental Table 10). Although the median genomic distance spanned by caPeak sets was ∼10 kb, many coordinated sets were much broader, and 271 sets spanned >100 kb (Fig. 2C; Supplemental Table 11).
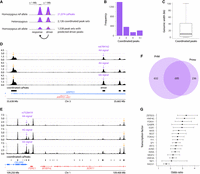
caQTL signals associated with multiple, coordinated peaks. (A) caQTL mapping strategy testing variants within 1 Mb of ATAC peak centers. A variant may affect the accessibility of a distal peak if it affects accessibility of the overlapping proximal “driver” peak, and the proximal peak affects the distal “response” peak. (B) Number of coordinated caPeak sets containing different numbers of caPeaks. (C) Distribution of genomic width covered by peaks in the coordinated sets. (D) Coordinated peaks at ARPP21. The predicted driver peak is indicated with the purple arrow, and the ARPP21 promoter peak in the coordinated set is indicated with the orange arrow. The ATAC signal tracks represent read depth–normalized ATAC signal averaged across individuals for the three genotypes of caQTL lead variant rs6784162 (purple arrow), which is the lead variant in the 1 kb and 1 Mb caQTL analyses. (E) Coordinated peaks at SORT1 with predicted driver peak (purple arrow) and one of the SORT1 promoter peaks (orange arrow). Read depth–normalized ATAC signal tracks averaged by genotype for rs7528419, which is the lead variant in the 1 kb and 1 Mb caQTL analyses. The previously described functional variant at this locus, rs12740374, is located in the same peak. (F) Overlap of driver peaks predicted by proxy overlap and PHM. (G) Enrichment of TF ChIP-seq binding sites from ENCODE in driver peaks relative to response peaks. Odds ratios and 95% confidence intervals. All TFs are significant at a P-value threshold of 2.9 × 10−3.
We identified 418 sets of coordinated peaks that contained both promoter and distal peaks (Supplemental Table 9). At one example near ARPP21, a set of four coordinated peaks spanning ∼26 kb consisted of a promoter peak for ARPP21 and three distal peaks within the ARPP21 gene body (Fig. 2D). At the well-characterized SORT1 locus (Musunuru et al. 2010), a set of 28 coordinated peaks spanning ∼143 kb contained five promoter peaks (three for SORT1, one for CELSR2, and one for MYBPHL) (Fig. 2E).
We hypothesized that many sets of coordinated peaks would contain a single “driver” peak responsible for the primary caQTLs association that regulates the other “response” peaks. We used two complementary approaches to predict driver peaks: proxy overlap, which predicts a driver peak if only one peak in a coordinated set overlaps proxy variants of the caQTL signal, and a pairwise hierarchical model (PHM) (Kumasaka et al. 2019), which is a Bayesian model that predicts causal interactions between pairs of peaks. We identified a single driver peak for 931 coordinated peak sets using proxy overlap and 1348 sets using PHM (Supplemental Table 9). The methods showed strong overlap: 695 (75%) of the 931 proxy overlap driver peaks were also identified by PHM (OR = 2.9, P = 1.5 × 10−29, two-sided Fisher's exact test) (Fig. 2F), including the example near ARPP21 (Fig. 2D). At the SORT1 locus, proxy overlap was not able to identify a single driver peak because two caPeaks overlapped proxy variants. However, PHM was able to predict a single driver peak that overlapped a previously described functional variant (rs12740374, LD r2 = 1 with caQTL lead, TOPMed Europeans) (Musunuru et al. 2010). In the vast majority of cases, both methods identified the caPeak with the most significant caQTL P-value at the signal as the driver peak (proxy overlap, 84%; PHM, 89%) (Supplemental Table 9). In total, we identified a driver peak for 1538 coordinated sets, selecting the proxy overlap peak if the methods disagreed.
To determine if driver peaks had distinct regulatory characteristics from response peaks, we examined binding to specific TFs. Driver peaks were significantly more likely (OR > 1, P < 2.9 × 10−3) to overlap ChIP-seq binding sites of all 17 tested TFs (Ramaker et al. 2017; The ENCODE Project Consortium et al. 2020), including the key liver regulators HNF4A, HNF4G, and FOXA1/2 (Fig. 2G; Supplemental Table 12). However, only one TF motif, for HNF4A, was enriched in driver peaks (OR > 1, P < 1.8 × 10−4) (Supplemental Table 13). Taken together, these results show that driver peaks are more likely to overlap binding sites of multiple TFs, suggesting that they may be crucial regulatory focal points at coordinated caQTL signals.
Identifying target genes for caPeaks
We used four approaches to link caPeaks to putative target genes (Fig. 3A; Supplemental Tables 9, 14–17): proximity to transcription start site (TSS; promoter) from GENCODE (Fig. 3B; Frankish et al. 2019), colocalization of caQTLs and liver tissue eQTLs from GTEx (Fig. 3B; The GTEx Consortium 2020), chromatin interactions from promoter capture Hi-C from liver tissue (Jung et al. 2019) and HepG2 (Fig. 3C; Chesi et al. 2019; Selvarajan et al. 2021), and coordination of caPeaks with promoter caPeaks (Fig. 2D,E). For 82% of colocalized caQTL and eQTL signals, the alleles associated with higher accessibility were also associated with higher gene expression, similar to previous findings that most colocalized caQTLs and eQTLs showed the same direction of effect (Degner et al. 2012; Currin et al. 2021); the remaining 18% of caQTL–eQTL colocalizations may represent transcriptional silencers. Among nonpromoter caQTLs, caPeaks linked by Hi-C were further from the gene TSS compared with caPeaks linked by eQTLs or coordination with promoter peaks (Fig. 3D; Supplemental Fig. 4; Supplemental Table 17). Only 965 of the 16,198 caPeak–gene links were supported by two or more approaches, consistent with the dependence of the methods on distance and demonstrating the utility of using multiple approaches. We identified caPeak links to 550 unique genes involved in drug response (Supplemental Table 17). Using the Human Protein Atlas (Uhlén et al. 2015), we identified 469 target genes whose expression was enriched in liver tissue (Fig. 3E; Supplemental Table 17), including HMGCL and CDO1 (Fig. 3B,C). Relative to genes not linked to caPeaks, caPeak target genes were more likely to be enriched in liver (7.7% vs. 3.9%, OR = 2.1, P < 3.2 × 10−29) (Supplemental Table 18).
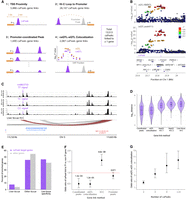
Predicted target genes of caPeaks. (A) Four methods used to link caPeaks to genes: transcription start site (TSS) proximity, promoter capture Hi-C from HepG2 and liver tissue, coordination of caQTLs for distal and promoter peaks, and colocalization of caQTLs with liver tissue eQTLs. (B) A HMGCL TSS-proximal caPeak (peak4713) for a caQTL colocalized with an HMGCL eQTL. The caQTL lead variant (rs58035855) is shown in each plot by a purple diamond, and colors represent linkage disequilibrium (LD) r2 values from 1000G Europeans. Gray points represent variants that were not in the 1000G LD reference panel. (C) A caPeak linked to CDO1 by Hi-C. Read depth–normalized ATAC signal tracks averaged by genotype for rs6863733, which is the lead variant in the 1 kb and 1 Mb caQTL analyses. (D) The distribution of log10 distances between caPeaks and the 5′-most TSS of linked genes. “HepG2 Hi-C 1” and “HepG2 Hi-C 2” indicate Hi-C results from two independent studies. (E) Percentage of caPeak target genes (purple) and non-caPeak genes (gray) enriched in liver tissue or other tissues. The non-caPeak target genes are any gene detected in the Human Protein Atlas that was not linked to a caPeak. (F) Logistic regression results testing if links between peaks and TFs are more likely to be supported by each of the linking methods relative to non-TF genes. Odds ratios and 95% confidence intervals, with P-values above. (G) Logistic regression results testing if coordinated caQTLs are more likely than noncoordinated caQTLs to colocalize with GTEx liver tissue eQTLs. Odds ratios and 95% confidence intervals for enrichment using sets of coordinated caQTLs with exactly two peaks, exactly three peaks, exactly four peaks, and five or more peaks.
Because of the role of TFs in gene regulation, we investigated the 663 links between 590 unique caPeaks and genes encoding 384 unique TFs. Compared with links between caPeaks and non-TF genes, links between caPeaks and TF genes were less likely to be supported by eQTLs (OR = 0.49, P = 1.2 × 10−5) and were more likely to be supported by Hi-C (OR = 1.57, P = 4.0 × 10−8) (Fig. 3F; Supplemental Table 19). caPeaks linked to TFs were generally farther from the TSS of the linked gene compared with caPeaks linked to non-TF genes (one-sided Mann–Whitney U test, P = 2.2 × 10−10 using the 5′-most TSS for the gene) (Supplemental Table 20), but we still observed a depletion of eQTL support for caPeak–TF links when adjusting for the distance to the TSS (Supplemental Table 19). We confirmed these findings when restricting to genes tested for eQTLs in GTEx liver (Supplemental Table 19). Among genes linked to caPeaks, TF genes showed a trend toward lower expression compared with non-TF genes (one-sided Mann–Whitney U test, P = 0.086) (Supplemental Fig. 5). However, when adjusting for gene expression level, we still observed that caPeak links to TF genes were depleted for eQTLs and enriched for Hi-C (Supplemental Table 19). These results suggest that caPeaks may have an overall weaker effect on expression of TFs relative to non-TF genes and that these weaker effects are less likely to be detected as eQTLs in current sample sizes.
Reasoning that variants with widespread effects on chromatin accessibility may be more likely to alter gene expression, we compared eQTL colocalization rates between coordinated and noncoordinated caQTLs. Coordinated caQTLs, including those without promoter peaks, were more likely than noncoordinated caQTLs to colocalize with eQTLs (OR = 4.1, P = 2.9 × 10−93) (Fig. 3G; Supplemental Table 21), even after adjusting for TSS proximity (OR = 3.7, P = 9.0 × 10−79). We also found that relative to noncoordinated caQTLs, the OR of colocalization between coordinated caQTLs and eQTLs increases as the number of caPeaks associated with the caQTL increases (Fig. 3G; Supplemental Table 21). We observed similar results when using only the 931 driver peaks from proxy overlap instead of all coordinated peaks in a set, demonstrating that these results are not an artifact of colocalizing with multiple peaks per coordinated set. We also observed similar results when using the proxy overlap driver peaks and adjusting for the absolute value of caQTL effect size (Supplemental Table 21), indicating that our results were not owing to differences in caQTL strength between coordinated and noncoordinated peaks. These results suggest that genetic variants are more likely to affect gene expression if they alter the activity of multiple regulatory elements than if they alter the activity of just one element, supporting previous findings (Kumasaka et al. 2016).
caQTL heritability enrichment and GWAS colocalization
We next identified GWAS traits that showed heritability enrichment in liver caPeaks among 703 traits from the UK Biobank (UKBB). Traits relevant to liver function showed significant heritability enrichment in caPeaks (FDR < 5% calculated from LDSC coefficient P-value) (Fig. 4A; Supplemental Fig. 6; Supplemental Table 22), including plasma levels of liver enzymes, proteins produced by the liver (albumin, sex hormone binding globulin [SHBG], and C-reactive protein [CRP]), and lipid traits. Our results are consistent with previously identified heritability enrichment for plasma levels of cholesterol/lipid traits and liver enzymes in liver regulatory elements (Finucane et al. 2015; Currin et al. 2021).
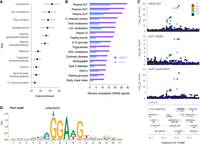
Heritability enrichment and GWAS colocalization. (A) Heritability enrichment of GWAS traits in caPeaks (FDR < 5%). Enrichment fold change and standard errors. (B) Percentage of GWAS signals for each trait that is colocalized with caQTLs (purple) or eQTLs (blue). (C) Colocalized GWAS (gamma-glutamyltransferase), eQTL (TGFB1), and caQTL (caPeak156441) associations. The caQTL lead variant (rs73045269) is shown in each plot by a purple diamond, and colors represent LD r2 values from 1000G Europeans. (D) Sequence logo motif for FLI1 that is disrupted by caQTL variant rs56254331 (position shown by arrow) within caPeak156441. The motif match is on the negative strand, so the C allele of rs56254331 corresponds to G in the motif. The C allele (G in the motif) is predicted to have a stronger motif match compared with the A allele (T in the motif).
To predict functional variants and effector regulatory elements at GWAS signals, we tested for colocalization between caQTLs and GWAS signals for cardiometabolic and/or liver function traits. Plasma levels of liver enzymes, cholesterol traits, CRP, and vitamin D had relatively high percentages of colocalized signals compared with other tested traits (Fig. 4B; Supplemental Table 23), consistent with the heritability enrichment of these traits in caPeaks (Fig. 4A). After accounting for coordinated peaks, the caQTL–GWAS colocalizations consisted of 844 unique caQTL signals and 998 unique caPeaks. We estimate that 673 of 4330 (16%) LD-clumped GWAS signals (r2 > 0.5) across all traits are colocalized with caQTLs (Supplemental Table 24). At colocalized signals containing coordinated caQTLs with predicted driver peaks, using the predicted driver peak for a set reduced the number of candidate functional peaks from 228 to 105, suggesting that identifying driver peaks helps fine-map variants and regulatory elements at GWAS signals.
We tested if caQTLs would identify more colocalized GWAS signals compared with an eQTL study of similar sample size. For all tested traits, more GWAS signals were colocalized with caQTLs compared with GTEx eQTLs in 178 samples (Fig. 4B; Supplemental Table 24). Among cross-trait clumped GWAS signals, 2.8-fold more were colocalized with caQTLs (673) than with eQTLs (239) (Supplemental Table 24). We confirmed that more GWAS signals were colocalized with caQTLs from the 1 Mb analysis compared with eQTLs (Supplemental Table 24), indicating that this result was not owing to the decreased multiple testing burden in the 1 kb caQTL analysis. The greater GWAS colocalization with caQTLs than with eQTLs is consistent with a more direct effect of genetic variants on chromatin accessibility than gene expression.
Regulatory mechanisms at GWAS signals
At caQTL signals colocalized with GWAS signals, we used predicted caPeak target genes and disrupted TF motifs to predict regulatory mechanisms. Of the 998 unique caPeaks with a GWAS colocalization, 487 had a predicted target gene, 462 contained a disrupted motif, and 226 had both target genes and disrupted motifs (Supplemental Table 23). We identified 231 caQTL signals that colocalized with both GWAS and eQTL signals, consistent with joint genetic regulation of chromatin accessibility, gene expression, and a GWAS trait. At an example near TGFB1, a caQTL for caPeak156441 is colocalized with an eQTL for TGFB1 and GWAS signals for levels of gamma-glutamyltransferase (GGT) (Fig. 4C), high-density lipoprotein (HDL) cholesterol, and coronary artery disease. Variant rs56254331 within the peak disrupts five TF motifs, with the strongest disruption for FLI1 (motif match on the antisense strand) (Fig. 4D; Supplemental Table 23). Three of these motifs were predicted to be associated with chromatin accessibility (Supplemental Table 8), and these three motifs all match ETS family TFs. For all three motifs, the allele that matches the motif better is associated with higher chromatin accessibility (Supplemental Data), higher expression of TGFB1, and higher levels of all three GWAS traits (Supplemental Table 23), suggesting that an ETS family TF may bind to the regulatory element marked by caPeak156441 to increase expression of TGFB1. At 228 of the GWAS-colocalized caQTL signals, the caPeak was linked to a gene, but neither the caQTL nor the GWAS signal was colocalized with an eQTL, demonstrating that caQTLs can be combined with other lines of evidence to predict target genes at GWAS signals that may be missed by eQTL data sets.
Functional validation of regulatory activity at GWAS loci
We sought to experimentally validate regulatory activity at selected colocalized caQTL and GWAS signals. First, at a GGT GWAS signal near TENM2, we examined a caQTL that consisted of the largest set of coordinated caPeaks colocalized with any GWAS signal (Fig. 5A,B). The 40 coordinated caPeaks spanned ∼1.2 Mb and contained promoter peaks for TENM2 (Fig. 5A,C). The caQTL signal colocalized with eQTL signals for TENM2 and a noncoding intronic transcript (Fig. 5B). The caQTL lead variant rs7726117 was the only variant at the signal (LD r2 > 0.8) that overlapped a caPeak, which was the driver peak (peak273749). We tested 329 bp DNA elements containing each allele of rs7726117 (Supplemental Table 25) for allelic differences in transcriptional activity using luciferase reporter assays in HepG2 cells (López-Terrada et al. 2009). The element containing the rs7726117-A allele showed significantly higher transcriptional activity relative to the element containing the C allele in both the forward (fold change [FC] = 1.5, P < 1 × 10−4) and reverse (FC = 1.3, P < 1 × 10−4) orientations relative to a minimal promoter (Fig. 5D; Supplemental Fig. 7). The rs7726117-A allele is also associated with increased chromatin accessibility, increased gene expression, and decreased GGT levels in the GWAS data (Fig. 5E).
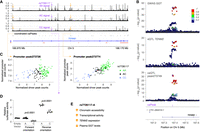
Functional validation at TENM2. (A) Coordinated peaks at TENM2 with driver caPeak273749 (purple arrow) and two promoter peaks for TENM2 (orange arrows). Read depth–normalized ATAC signal tracks averaged by genotype for rs7726117, which is the lead variant in the 1 kb and 1 Mb caQTL analyses. (B) Colocalized GWAS (GGT), eQTL (TENM2), and caQTL (caPeak273749) associations. The caQTL lead variant (rs7726117) is indicated in each plot by a purple diamond, and colors represent LD r2 values from 1000G Europeans. (C) Correlation of driver peak counts with counts of two TENM2 promoter peaks: peak273726 (the 5′-most promoter) and peak273774 (the more-downstream promoter). Points are colored by the genotype of rs7726117. Peak counts were normalized by the covariates used for caQTL mapping: sex, two genotype PCs, and 25 ATAC PCs. (D) Transcriptional activity in HepG2 cells of a 329 bp DNA element spanning caPeak273749 and containing rs7726117. The DNA element was tested in both orientations relative to the genome. (EV) Empty vector. Symbols represent two independently transfected wells for each of four independent clones for each allele; bars indicate mean and standard deviation; P-values are from t-tests of allelic differences. (E) Predicted mechanism at the TENM2 locus. The rs7726117-A allele shows higher transcriptional activity and is associated with greater liver chromatin accessibility, higher liver expression of TENM2, and lower plasma levels of gamma-glutamyltransferase.
Second, at GWAS signals for GGT and HDL cholesterol near RALGPS2, we examined a caQTL for a set of eight coordinated caPeaks that colocalized with an eQTL for RALGPS2 (Fig. 6A,B). The predicted driver peak at this signal is caPeak21014, which is linked to the promoters of RALGPS2, RALGPS2-AS1, and ABL2 by promoter capture Hi-C. Three variants at this caQTL signal overlap the driver peak, including the lead variant rs17361251 and two proxies (LD r2 = 1), rs17276513 and rs17276527. The AAA haplotype is associated with higher chromatin accessibility and gene expression. Using 323 bp DNA elements containing the three-variant haplotypes (Supplemental Table 25), we observed significantly higher transcriptional activity for the AAA haplotype than for the CTG haplotype in both the forward (FC = 1.6, P < 1 × 10−4) and reverse (FC = 1.9, P < 1 × 10−4) orientations relative to a minimal promoter (Fig. 6C,D; Supplemental Fig. 8).
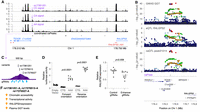
Functional validation at RALGPS2. (A) Coordinated peaks at RALGPS2 with driver caPeak21014 (purple arrow). Read depth–normalized ATAC signal tracks averaged by genotype for rs17361251, which is the lead variant in the 1 kb caQTL analysis and is in very strong LD (r2 = 0.998, TOPMed Europeans) with the 1 Mb lead (rs6701606). (B) Colocalized GWAS (GGT), eQTL (RALGPS2), and caQTL (peak21014) associations. The caQTL lead variant (rs17361251) is indicated in each plot by a purple diamond, and colors represent LD r2 values from 1000G Europeans. (C) Study design showing the location of the three variants within caPeak21014 that were tested in a haplotype for luciferase activity (for results, see D) and positions of gRNAs for CRISPRi (blue) relative to the peak (for results, see E). (D) Transcriptional activity in HepG2 cells of a 323 bp DNA element spanning caPeak21014 and containing rs17361251, rs17276513, and rs17276527. The DNA element was tested in both orientations relative to the genome. (EV) Empty vector. Symbols represent the average of two transfected wells for each of eight independent clones for each haplotype; bars indicate mean and standard deviation; P-values are from t-tests of haplotype differences. (E) Gene expression measured after CRISPRi of the caPeak21014 region. Compared with a pool of nontargeting control gRNAs, a pool of gRNAs targeted to the enhancer led to lower expression of RALGPS2. Each point represents the mean of three qPCR replicates for an independently transfected well. Lines indicate the mean of the 10–11 wells; P-value is from a t-test. (F) Predicted mechanism at the RALGPS2 locus. The AAA haplotype of variants rs17361251, rs17276513, and rs17276527 showed higher transcriptional activity and is associated with higher liver chromatin accessibility, higher liver expression of RALGPS2, and higher plasma levels of gamma-glutamyltransferase.
To validate if caPeak21014 regulates RALGPS2, we used CRISPRi to repress the peak region in HepG2 cells and measured expression of RALGPS2. Cells with guide RNAs (gRNAs) targeting caPeak21014 showed a 26% reduction in RALGPS2 expression compared with cells with nontargeting gRNAs (P < 8 × 10−3) (Fig. 6C,E; Supplemental Table 26). These results indicate that caPeak21014 is an enhancer for RALGPS2 and that interfering with caPeak21014 decreased RALGPS2 expression, which is consistent with the lower accessibility of the GTC haplotype and lower expression in the QTL data. In summary, we identified functional variants at this GWAS signal for GGT and HDL and showed that these variants may regulate expression of RALGPS2 by altering accessibility of caPeak21014.
Discussion
Here, we identified 35,361 caQTLs in liver tissue, 10-fold more than previously identified, including 156 caQTLs detected only in non-European study participants. We identified 2126 caQTLs associated with multiple, coordinated regulatory elements, including 1538 sets with predicted driver peaks. The caPeaks were enriched for heritability of liver-relevant traits, and 844 caQTL signals colocalized with GWAS signals, suggesting specific variants that may be responsible for the shared signals. We identified caPeak target genes and disrupted TF motifs to predict mechanisms at GWAS signals and validated regulatory effects at two loci. Together, these results demonstrate the utility of well-powered caQTL studies to understand long-range gene regulation and identify regulatory mechanisms at GWAS loci.
Mapping caQTLs in even relatively modest sample sizes is useful to predict regulatory mechanisms at GWAS signals. Despite a slightly smaller sample size for liver caQTLs than for liver eQTLs (The GTEx Consortium 2020), more GWAS signals colocalized with caQTLs than with eQTLs. For 228 caQTL signals that colocalized with GWAS but not eQTLs, other methods could link the caPeak to a target gene. Generally, caQTLs could have larger effect sizes than eQTLs (Liang et al. 2021), especially if variants more directly alter regulatory element accessibility than gene expression. Some signals lacking detectable eQTLs may have effects on expression that are too small to detect in the available samples. A recent study reported that genes near GWAS signals are under strong selective pressure and may be harder to detect in eQTL studies (Mostafavi et al. 2023). In a model in which variants affect complex traits by altering transcriptional regulation, we expect that caQTL effect sizes would be larger than eQTL effect sizes and that eQTL effect sizes would be stronger than GWAS effect sizes. However, many GWAS studies analyzed here had sample sizes in the hundreds of thousands and were much more powered to detect variants with small effect sizes than eQTL studies with sample sizes of a few hundred. Therefore, detection of GWAS–caQTL colocalizations without an eQTL could result from large caQTL effect sizes detectible in relatively small caQTL sample sizes, smaller eQTL effect sizes not detectible in current eQTL sample sizes, and even smaller GWAS effect sizes detectible in large GWAS sample sizes. Outside of sample size considerations, other caQTL signals without colocalized eQTL might represent primed enhancers near context-specific genes for which the caQTL is detectable before exposure but for which the eQTL is only detectable after exposure (Alasoo et al. 2018). Larger caQTL studies in additional tissue, cell, and exposure contexts should provide power to predict even more regulatory mechanisms.
We identified and characterized caQTL signals associated with multiple elements that are presumably coordinately regulated. Similar to findings from studies in other tissues (Grubert et al. 2015; Waszak et al. 2015; Chen et al. 2016; Kumasaka et al. 2016, 2019; Alasoo et al. 2018; Gate et al. 2018; Delaneau et al. 2019), we found that caQTL signals that contain multiple elements are numerous and can span hundreds of kilobases. Although we and others identified coordinated elements by grouping elements that shared the same caQTL signal (Grubert et al. 2015; Alasoo et al. 2018; Gate et al. 2018), other studies used alternative approaches; Waszak et al. (2015) identified sets of correlated elements, called variable chromatin modules (VCMs), and then tested for QTLs using the first eigenvalue of these VCMs, and Kumasaka et al. (2019) used PHM to map pairs of interacting elements. We compared two methods for predicting driver elements and found ∼75% of proxy overlap drivers were shared with PHM, which demonstrates that the two methods may perform better in different situations. Proxy overlap is useful when only one caPeak overlaps proxy variants, but PHM can help pinpoint the driver when multiple caPeaks overlap proxies. It is also possible that some signals may contain more than one driver element or that some represent pleiotropic associations that are not causally related. We contributed to the mechanistic understanding of genetically coordinated elements by showing that driver elements were more likely than response elements to overlap binding sites of several TFs, although this analysis was limited to 17 TFs with available liver tissue ChIP-seq in ENCODE (Ramaker et al. 2017; The ENCODE Project Consortium et al. 2020). The binding sites of these TFs in liver have been shown to have a high degree of overlap (Ramaker et al. 2017), which explains why driver peak enrichment results were similar between the TFs. Motif enrichment analyses only identified one TF motif enriched in driver elements relative to response elements, which could be explained by the presence of suboptimal motifs in enhancers missed by our analysis (Crocker et al. 2015; Dror et al. 2015; Farley et al. 2015) or by indirect binding mediated by cooperation with sequence-bound TFs (Gordân et al. 2009). The enrichment of a motif for HNF4A suggests that this TF may be crucial in establishing accessible chromatin and subsequent binding by other TFs at driver elements.
The peak–gene links identified by different approaches had different characteristics. Coordinated regulation and eQTL colocalization tended to identify links for peaks that were more TSS proximal, whereas promoter capture Hi-C tended to link peaks that were more distal. However, the discovery of colocalized eQTLs was limited by eQTL study power, causing more distal conditionally distinct eQTLs to be missed (Dobbyn et al. 2018; Raulerson et al. 2019; Brotman et al. 2025), and Hi-C restriction fragment resolution and the removal of artifacts from potential self-ligation limited detection of peak–gene links at short distances. We also found that Hi-C was more likely than eQTLs or coordinate regulation to link peaks to genes encoding TFs. Previous studies found that TFs are less likely to have eQTLs compared to non-TF genes (Battle et al. 2014; Mostafavi et al. 2023). Generally, eQTL–caQTL colocalization likely provides the strongest evidence that a peak is linked to a gene because both accessibility and expression levels change with genotype, although eQTL detection is limited by sample size. Coordinated change in the accessibility of distal and promoter peaks provides relatively strong evidence linking peaks to genes, but detecting long-range caQTLs is limited by sample size, and a distal enhancer does not necessarily have to alter promoter accessibility to affect a gene. TSS proximity and Hi-C interactions are both useful for identifying more peak–gene links at smaller sample sizes, but simple proximity to the TSS in linear or 3D space provides a weaker form of evidence. Overall, using multiple approaches allows more comprehensive detection of target genes.
Our functional assays supported predictions of regulatory mechanisms at two loci. At TENM2, we showed that rs7726117 exhibited allelic differences in transcriptional reporter activity consistent with the TENM2 eQTL (The GTEx Consortium 2020). We did not attempt to validate the link between caPeak273749 and TENM2 using CRISPRi in HepG2 because, based on ENCODE RNA-seq and ATAC-seq data (The ENCODE Project Consortium et al. 2020), TENM2 expression and caPeak273749 accessibility were low in the HepG2 model. Other approaches or cell models are needed to validate that regulatory element-gene link. At RALGPS2, we were able to validate allelic differences in transcriptional activity and the peak–gene link. Interfering with caPeak21014 using CRISPRi did not completely knockdown RALGPS2, which could indicate that RALGPS2 is regulated by additional elements, that the accessibility of the peak was not completely attenuated, or that the assay was not optimized. Further validation of these and other predicted mechanisms is needed.
Although the current study made a substantial contribution to our understanding of genetic effects on regulatory elements, it was limited to a single tissue, and most individuals were genetically similar to Europeans. Further, we did not map caQTLs on sex chromosomes. Well-powered caQTL studies in additional tissues are needed to identify tissue-specific genetic effects on regulatory elements, and single-cell studies are needed to identify the relevant cell types for caQTLs in heterogeneous tissues. Our results also show that caQTLs can vary across populations owing to allele frequency differences, but identification of population-specific caQTLs was limited by sample size in our study; caQTL mapping in larger, more diverse cohorts may identify more caQTLs that differ across populations. Large-scale functional assays will help confirm links between variants, regulatory elements, and genes predicted by caQTL studies.
Methods
Ethics statement and liver tissue samples
Noncancerous liver tissue samples from deceased, deidentified human donors were previously obtained (Innocenti et al. 2011), and use of these liver tissue samples in the current study was considered to be non-human subject research by the institutional review boards at St. Jude Children's Research Hospital (Memphis, TN) and the University of North Carolina (Chapel Hill, NC). Cause of death was recorded for all but eight participants. The most common causes of death included cerebrovascular accident, head trauma, and anoxia.
Nuclei extraction and ATAC-seq library preparation
Nuclei were extracted from frozen liver tissue samples as previously described (Currin et al. 2021). We prepared ATAC-seq libraries using the Omni-ATAC protocol (Corces et al. 2017) with 5 µL Tn5 transposase per reaction, cleaned the transposase reaction and final libraries with Zymo DNA Clean & Concentrator (D4029), visualized and quantified the libraries using a TapeStation, and sequenced 150 bp paired-end reads on either an Illumina HiSeq 4000 or HiSeq X. We initially generated three ATAC-seq libraries and aimed for 75 million high-quality aligned read pairs per individual. We generated additional libraries and/or resequenced existing libraries to increase depth for some individuals (Supplemental Tables 2, 3). After quality control, the number of libraries per individual ranged from one to six, with most individuals having either three or four libraries.
ATAC-seq read alignment and quality control
We trimmed sequencing adapters and aligned ATAC-seq reads to the GRCh38 build of the human genome (International Human Genome Sequencing Consortium 2001) and generated high-quality (mapq > 20), nonduplicated alignments as previously described (Perrin et al. 2021). We converted BAM files to BED files using BEDTools (Quinlan 2014) and called peaks on each ATAC-seq library separately using MACS2 (Zhang et al. 2008) with the parameters -q 0.05 –nomodel –shift -100 –extsize 200 –keep-dup all. We removed ATAC libraries with fewer than 10 million raw read pairs, TSS enrichment less than four as calculated by ATAQV (Orchard et al. 2020), and <10% of high-quality alignments overlapping peaks from downstream analyses. We also removed two libraries that were identified as outliers using PCA of normalized read counts overlapping a previously defined set of liver ATAC peaks (see Supplemental Methods; Currin et al. 2021).
Validation of sample identity
Genotyping for 224 liver donors was previously described (Innocenti et al. 2011). We obtained genotypes from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE26105. We tested for sample swaps between ATAC-seq and genotype data using verifyBamID (Jun et al. 2012) using variants overlapping existing liver ATAC peaks (see Supplemental Methods; Currin et al. 2021). We considered the best-matching ATAC library to a set of genotypes to be a confident match if chipmix <0.02. We corrected sample swaps and removed ATAC libraries from further analysis if they did not confidently match to genotypes from any of the 224 individuals.
Genotype quality control
We used KING v2.2.5 (Manichaikul et al. 2010) with the parameters –unrelated –degree 2 to remove highly related individuals with up to second-degree relatedness. Using PLINK v1.9 (Purcell et al. 2007), we removed genotypes with missingness across samples >5% and excluded individuals that had genotype missingness >5%, heterozygosity F coefficient greater than four standard deviations from the mean, and/or inconsistency between reported sex and sex estimated from genotypes (see Supplemental Methods).
Selection of the final sample set and identification of consensus ATAC peaks
After ATAC and genotype quality control, 477 ATAC libraries from 138 individuals remained. For each individual, we merged the BAM files of the high-quality libraries and removed duplicates using Picard MarkDuplicates (https://github.com/broadinstitute/picard) because some libraries for the same donor represent resequencing of the same library preparation. We then called peaks on merged libraries per individual using the same procedure that we used for single libraries (see section “ATAC-seq read alignment and quality control”).
We merged peaks across individuals using BEDTools (Quinlan 2014) and retained merged peaks found in at least seven individuals (5% of 138). To verify the reported sex of the ATAC libraries, we counted the number of ATAC reads overlapping merged peaks using featureCounts (Liao et al. 2014), calculated the proportion of sex chromosome peak counts found on the X Chromosome (Chr X counts/[Chr X counts + Chr Y counts]), and classified a sample as female if >99% of counts on the sex chromosomes were found on the X Chromosome and as male otherwise.
Merging peaks across individuals can result in wide peak boundaries and concatenation of multiple nearby accessible chromatin regions into one peak. Therefore, we developed a procedure to refine the boundaries of the merged peaks found in at least seven individuals. We first calculated the number of individuals that had a peak overlapping each base of each merged peak, which we define as coverage, using BEDTools coverage (Quinlan 2014). We converted the output of BEDTools coverage into a matrix of coverage values with peak IDs as rows and peak bases as columns. For each peak, we identified all local minima in coverage; peaks that contain only one peak should have only two local minima, whereas peaks containing multiple subpeaks will have three or more local minima. To prevent classifying small fluctuations in coverage as subpeaks, we only retained adjacent pairs of minima if the distance between them was at least 200 bp and if the maximum coverage between them was both at least seven and greater than twice the maximum of the two minima. To define consensus peak boundaries, we identified the position between the two local minima with maximum coverage and extended in both directions until we reached the outermost positions that had half of the maximum coverage. This is similar to the full width at half maximum procedure used to identify boundaries of DNase peaks in ENCODE (Meuleman et al. 2020).
We determined which consensus peaks were shared with ATAC peaks from our pilot liver caQTLs study (Currin et al. 2021) and liver tissue ATAC peaks from ENCODE (The ENCODE Project Consortium et al. 2020) using BEDTools (see Supplemental Methods; Quinlan 2014).
Genotype imputation
We performed genotype imputation for 157 individuals that passed genotype quality control and that had ATAC data using the TOPMed imputation server (Das et al. 2016; Taliun et al. 2021), selecting Eagle v2.4 (Loh et al. 2016) for phasing and Minimac4 v1.0.2 (Das et al. 2016) for imputation (see Supplemental Methods). We filtered the imputation results to contain only the 138 individuals that passed ATAC quality control and biallelic single-nucleotide polymorphisms (SNPs) or indels with imputation r2 > 0.3 and minor allele count ≥ 10 using BCFtools v1.13 (Danecek et al. 2021).
Ancestry classifications and genotype principal components
We generated a VCF file containing the 138 liver donors and the 1000 Genomes (1000G) individuals and that contained 99,206 common variants in approximate linkage equilibrium (see Supplemental Methods). We performed two rounds of genotype PCA using PLINK (Purcell et al. 2007): one using only the 138 liver donors to use for QTL covariates and one using both the liver donors and 1000G individuals to use for ancestry inference. We used two methods to infer ancestry: weighted k-nearest neighbors (WKNN), implemented in the kknn R (R Core Team 2015) package, and support vector machine (SVM), implemented in the e1071 R package. For both methods, we used the first two genotype PCs to calculate distances between individuals and predicted the ancestry of liver donors using models trained on 1000G individuals. For WKNN, we estimated the optimal value of k to be four using the train.kknn function. Both methods agreed that 118 individuals were similar to European populations, 17 individuals were similar to African populations, and one individual was similar to admixed American populations; we considered the remaining two individuals for which the methods disagreed as similar to admixed populations.
caQTL mapping
We removed ATAC mapping bias using the WASP mapping pipeline (van de Geijn et al. 2015), quantified peak accessibility using featureCounts (Liao et al. 2014), adjusted for GC bias using EDASeq (Risso et al. 2011), and adjusted for library size and performed variance stabilization using DESeq2 (Love et al. 2014). We used ATAC PCs to control for unmeasured sources of variation. We mapped caQTLs using inverse-normal-transformed, variance-stabilized peak counts and variants within 1 kb of peak centers using FastQTL (Ongen et al. 2016) with the parameters –permute 1000 –normal –window 1000 –seed 123456789. We included sex, two genotype PCs, and 25 ATAC PCs as covariates. We included two genotype PCs because the percentage of variance explained leveled off after the first two PCs. We chose a window of 1 kb from peak centers to focus on variants close to and within the associated peaks. We chose 25 ATAC PCs because it resulted in the first local maximum of the number of significant caPeaks. To identify ATAC peaks with a significant caQTL (caPeaks), we estimated the false-discovery rate (FDR) of the FastQTL beta-approximated P-values for the most strongly associated variant per peak using the Benjamini–Hochberg (BH) procedure (Benjamini and Hochberg 1995) and classified caPeaks with FDR < 5% as significant. To test for long-range caQTL effects, we also ran FastQTL using variants within 1 Mb of peak centers with the parameter –window 1000000, keeping all other parameters identical to the 1 kb analysis. We chose a window of 1 Mb from peak centers to capture interactions between peaks at a locus but still within a typical distance for cis regulation.
We determined how many caQTLs from our previous study (Currin et al. 2021) were replicated in the current study (see Supplemental Methods). To compare the percentages of caPeaks and non-caPeaks (FastQTL beta-adjusted P-value > 0.5) overlapping different types of regulatory elements, we downloaded chromatin states lifted over to GRCh38 from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015; https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/core_K27ac/jointModel/final/all_hg38lift.dense.browserFiles.tgz) and assigned ATAC peaks to liver tissue (epigenome ID E066) chromatin states as previously described (Currin et al. 2021).
To identify caQTL that may be specific to non-European populations, we selected caQTL for which the lead variant was not polymorphic (MAF = 0) in donors similar to European populations. We also compared population-level lead variant MAF in our cohort to MAF in TOPMed (Taliun et al. 2021) populations present in the TOP-LD (Huang et al. 2022) resource.
Downsampling of caQTL sequencing depth and sample size
For sequencing depth, we downsampled the WASP-filtered BAM files to 20%, 40%, 60%, and 80% of the original depth using SAMtools (Danecek et al. 2021) with the -s parameter. We used five technical replicates for each downsampled percentage (random number seeds ranging from one to five). For each replicate, we quantified accessibility of the consensus peaks identified from the full-depth data and performed caQTL mapping using the same pipeline used for the full-depth data, including the caQTL covariates from the full-depth data.
For sample size, we generated five replicate random subsets of the full sample set corresponding to 20% (n = 27), 40% (n = 55), 60% (n = 82), and 80% (n = 110) of total sample size using R (random number seeds ranging from one to five) (R Core Team 2015). For each replicate, we generated a subset of the full-sample, variance-stabilized, peak-by-sample matrix containing the downsampled sample list, recalculated ATAC PCs, and performed caQTL testing using the peaks and variants used for the full sample list and including sex, two genotype PCs, and a variable number of ATAC PCs as covariates. We tested ATAC PCs from 0% to 25% of the downsampled sample size and selected the number of PCs that resulted in the first maximum of the number of significant caPeaks.
Identification of TF motifs disrupted by caQTL variants
To identify caQTL variants that may disrupt TF binding, we searched for cases in which the strength of a TF motif match changed between the alleles of a caQTL variant. Although ChIP-seq is a more direct measure of TF binding than motifs, measuring allelic differences in binding using ChIP-seq is limited by the number of TFs with available ChIP-seq data, and allelic imbalance in ChIP-seq reads can only be measured at heterozygous sites with sufficient read depth in the respective data sets. Consequently, we used TF motifs to more comprehensively survey allelic effects of caQTL variants on TF binding. We tested if caQTL variants were more likely to disrupt specific motifs among a set of 516 motifs from Cis-BP v1.02 (Weirauch et al. 2014) using a previously described protocol (Currin et al. 2021) with a few changes. To ensure that the set of background non-caQTL variants was larger than the set of caQTL variants, we did not use propensity score matching when selecting non-caPeaks (FastQTL beta-adjusted P > 0.5). However, we still adjusted for peak GC content and distance to the closest protein coding TSS in the logistic regression model that tested for motif disruption associated with caQTL status. We did not include peak width because it was not correlated with caQTL status. We tested for association of caQTL status with motif disruption for the 280 motifs that remained after selecting motifs disrupted by at least 20 caQTL variants and selecting the single motif per TF that had the most disruptions. We considered a motif to be significantly associated with caQTL status if the logistic regression P-value was less than 1.8 × 10−4 (0.05/280 motifs).
Identification of coordinately regulated caPeaks
We conservatively grouped caPeaks from the 1 Mb caQTL analysis whose lead variants were identical or in very high LD with each other (r2 > 0.9, TOPMed Europeans) (Taliun et al. 2021; Huang et al. 2022). We defined distance between variants as the complement of LD values (1 – r2), performed single linkage hierarchical clustering using the hclust function in R (R Core Team 2015), and identified clusters as groups with a maximum distance of 0.1 (1–0.9).
Identification of driver peaks at coordinated caQTL signals
We used two complementary methods to predict driver peaks at coordinated caQTLs: proxy variant overlap and the PHM (Kumasaka et al. 2019), which tests for causal interactions between pairs of peaks. For the proxy variant overlap approach, we identified all proxy variants of all clumped lead variants of a caQTL signal (LD r2 ≥ 0.8) and overlapped these proxies with all caPeaks from the 1 Mb analysis. We classified a coordinated caQTL as having a single driver peak if only one caPeak within the coordinated set overlapped proxies of the signal and no caPeaks within or outside the coordinated set overlapped proxies of the signal. We ran PHM to test for causal interactions between pairs of caPeaks from the 1 Mb analysis. We edited the bayeslm1.sh and bayeslm2.sh scripts provided with PHM to increase the –window-size parameter from 500,000 to 1,000,000 to test caPeak pairs up to 1 Mb apart. We classified a pair of peaks as having a causal interaction if the posterior probability that either peak regulates the other peak was greater than 0.5. We determined the number and identity of caPeaks regulating (upstream) and regulated by (downstream) each caPeak. To predict driver peaks at coordinated caQTLs, we searched for causal interactions between all caPeaks in the coordinated set, as well as any caPeaks outside the set that overlapped proxies of the coordinated caQTL signal. We considered a coordinated caQTL to have a single driver peak if only one of the tested peaks had at least one downstream peak within the coordinated set and no upstream peaks inside or outside of the coordinated set. If the proxy variant overlap and PHM methods disagreed on the driver peak for a coordinated caQTL, we classified the proxy variant overlap peak as the driver peak.
We tested for significant overlap between driver peaks identified by the two methods using a two-sided Fisher's exact test. The contingency table for the Fisher's exact test consisted of the number of coordinated signals with driver peaks predicted by both methods, proxy overlap only, PHM only, and neither method. To ensure that each coordinated signal was represented once, we assigned the driver peak to proxy overlap for the 46 coordinated signals in which PHM and proxy overlap disagreed on the driver peak prediction. We confirmed that the overlap between the two methods was still significant when retaining the PHM predictions at these 46 discordant signals (OR = 2.7, P = 4.6 × 10−26).
Enrichment of TF ChIP-seq peaks and motifs
We downloaded a metadata file of liver TF ChIP-seq conservative IDR thresholded peaks from ENCODE (The ENCODE Project Consortium et al. 2020) and removed files that had any notes in the “Audit NOT_COMPLIANT” column. The resulting data set consisted of files for 17 TFs (Ramaker et al. 2017), 15 of which were mapped in two donors (ENCDO060AAA and ENCDO882mMZ); we merged ChIP-seq peak summits across multiple donors for the same TF. For driver peaks, we used the 1538 peaks from the combined set of PHM and proxy overlap predictions. For response peaks, we used the 3034 nondriver peaks from the 1538 coordinated sets with a predicted driver peak. We extracted the central 200 bases of driver and response peaks to mitigate enrichment biases owing to differences in peak width distributions between the driver and response peaks. We used BEDTools intersect (Quinlan 2014) to count the number of driver and response peaks overlapping the single base pair summits of TF ChIP-seq peaks separately per TF. We determined if driver peaks were significantly more likely to overlap ChIP-seq summits of each TF using a Fisher's exact test and classified enrichments with OR > 1 and P < 2.9 × 10−3 (0.05/17 TFs) as significant.
We tested for enrichment of 286 TF motifs in driver peaks relative to response peaks. We generated the set of 286 motifs by selecting a single motif per TF based on highest information content from the set of 516 motifs used in the caQTL motif disruption analysis. We scanned the central 200 bp of driver and response peaks for motif occurrences using FIMO (Grant et al. 2011) with the default P-value threshold of 1 × 10−4. We computed the number of unique driver and response peaks that overlapped each motif using BEDTools (Quinlan 2014), tested for enrichment using a Fisher's exact test, and classified motifs as significantly enriched in the driver peaks (OR > 1) or response peaks (OR < 1) if P < 1.8 × 10−4 (0.05/283 motifs with at least one occurrence in driver or response peaks).
Genome browser views of caQTL signals
We visualized the genome context of caQTL signals using Plotgardener v1.2.10 (Kramer et al. 2022). To make tracks of ATAC signal, we generated bedGraph files normalized for sample read depth using BEDTools genomecov (Quinlan 2014), converted the bedGraph files to bigWig files using UCSCTools (Hinrichs et al. 2006), imported the bigWig files into R with the Plotgardener readBigwig function (Kramer et al. 2022), and calculated the mean of signal across individuals for each genotype of the caQTL lead variant. We incorporated GENCODE v41 (Frankish et al. 2019) protein-coding and lncRNA genes into Plotgardener (see Supplemental Methods).
Predicting target genes of caPeaks
TSS proximity
We obtained transcript and gene coordinates from the GENCODE (Frankish et al. 2019) v41 comprehensive GTF file for protein-coding and lncRNA genes. For each gene, we retained the 5′-most TSS. We also retained additional TSSs for a gene if they were within 500 bp and on the same strand as a CAGE peak from FANTOM5 (Abugessaisa et al. 2017; https://fantom.gsc.riken.jp/5/datafiles/reprocessed/hg38_latest/extra/CAGE_peaks/hg38_fair+new_CAGE_peaks_phase1and2.bed.gz). We classified a caPeak as proximal to the TSS of a gene if it was within 2 kb upstream of or 1 kb downstream from any TSS of the gene, identified using BEDTools (Quinlan 2014).
Promoter capture Hi-C
We obtained promoter capture Hi-C data from three studies: one mapped in liver tissue (Jung et al. 2019) and two mapped in HepG2 cells (Chesi et al. 2019; Selvarajan et al. 2021). Hi-C data mapped in liver tissue (Jung et al. 2019) were filtered for significant interactions as previously described (Currin et al. 2021). Significant Hi-C interactions at four-fragment DpnII resolution for HepG2 data set 1 (Chesi et al. 2019) were provided by the authors upon request. Significant Hi-C interactions for HepG2 data set 2 (Selvarajan et al. 2021) were obtained from the paper. For all three studies, we converted the coordinates of the Hi-C interactions from GRCh37 to GRCh38 using a wrapper script around the liftOver tool (Hinrichs et al. 2006), removing interactions in which both ends did not successfully liftOver or in which either end mapped to a different chromosome in the new build. To generate a consistent set of bait fragment-to-gene assignments across the three studies, we assigned a bait fragment to a gene if it overlapped any TSS of a gene in our high-confidence TSS set (see subsection “TSS proximity”). We identified caPeaks that overlapped either end of a Hi-C interaction using BEDTools (Quinlan 2014) and linked caPeaks to the gene(s) on the opposite end. Similar to a previous study (Jung et al. 2019), we removed interactions in which the two fragments were <15 kb apart to remove potential unligated or self-ligated fragments, and we removed interactions with fragments >2 Mb apart to focus on local regulatory effects; we measured distance between innermost fragment edges.
Colocalization of caQTLs and eQTLs
We obtained liver tissue eQTLs from GTEx v8 (FDR < 5%) (The GTEx Consortium 2020) and restricted to protein-coding and lncRNA genes whose Ensembl IDs were found within GENCODE v41 (Frankish et al. 2019). We used eQTL lead variants from the multiancestry analysis (n = 208) but used summary statistics from the subset of donors genetically similar to European populations defined by GTEx (n = 178) for colocalization. For caQTLs, we used the lead variants and summary statistics from the full set of 138 individuals. We classified a caQTL signal as colocalized with an eQTL signal if the lead variants had LD r2 > 0.5 (TOPMed [Taliun et al. 2021] European-ancestry calculated by TOP-LD [Huang et al. 2022]) and the posterior probability of colocalization (PP H4) estimated by the coloc (Giambartolomei et al. 2014). The approximate Bayes factor method was >0.8.
Coordination between TSS-proximal and -distal caPeaks
For the 1 Mb caQTL analysis, we linked a TSS-distal caPeak (>2 kb upstream of and >1 kb downstream from the TSS) to a gene if the distal caPeak was coordinated with a TSS-proximal caPeak.
Filtering of peak–gene links
We removed 18,189 caPeak–gene links that were supported by Hi-C in HepG2 but not supported by any other approach because many of these links may be specific to the cancerous and/or immortalized conditions of the HepG2 cell line.
Identification of caPeak target gene categories
We used the Human Protein Atlas (Uhlén et al. 2015) to determine the tissue specificity of protein coding caPeak target genes (https://www.proteinatlas.org/download/proteinatlas.tsv.zip). We considered a gene to be enriched in liver tissue if the Protein Atlas considered the gene to be “tissue enriched,” “tissue enhanced,” or “group enriched” and if liver was one of the tissues in the group. We determined if caPeak target genes were more likely to be enriched in liver tissue relative to genes detected in the Protein Atlas that were not caPeak target genes using logistic regression. To identify target genes that are TFs, we selected TFs that had either a directly determined or inferred binding motif from Cis-BP v2.00 (Weirauch et al. 2014) and restricted to autosomal genes from GENCODE v41 (Frankish et al. 2019) by matching on Ensembl ID. We determined if caPeaks were more or less likely to be linked to TFs compared with non-TF genes by each of the four peak–gene links separately using logistic regression. For eQTL, we also ran an additional model providing the distance to the nearest TSS for the same gene as a covariate. Because not all genes linked to caPeaks were tested for liver eQTLs in GTEx, we reran the logistic regression models only using those genes. We also ran logistic regression models using gene expression level as a covariate. To compare gene expression levels between TF and non-TF genes, we used liver gene counts from GTEx (https://storage.googleapis.com/adult-gtex/bulk-gex/v8/rna-seq/counts-by-tissue/gene_reads_2017-06-05_v8_liver.gct.gz) and calculated counts per million (CPM) using edgeR (Robinson et al. 2010) with the trimmed mean of the m-values method to calculate library sizes. We defined the expression level of a gene as the median CPM across individuals. To identify caPeak target genes involved in drug response, we obtained a previously compiled list of genes involved in drug response (Etheridge et al. 2020) and restricted to autosomal genes present in GENCODE (Frankish et al. 2019) v41 with biotype of protein coding or lncRNA by matching on gene symbol.
Enrichment of coordinated peaks and eQTLs
We tested if caQTL signals with coordinated peaks were more likely to colocalize with GTEx liver eQTLs compared with caQTL signals for noncoordinated peaks using logistic regression. We tested additional models including promoter peak status or distance to nearest TSS as a covariate. For coordinated peaks, we considered the set to have a promoter peak if at least one peak was TSS proximal. We calculated TSS distance using the peak in the coordinated set closest to the nearest TSS. For most models, we considered a coordinated peak set to colocalize with an eQTL if the caQTL for at least one of the component peaks colocalized with an eQTL. As this approach could potentially overcount coordinated peak sets, we also tested models using the 931 driver peaks predicted from the proxy overlap method to test for eQTL colocalization, one peak per coordinated set. As the background peaks, we used noncoordinated caQTL peaks for which caQTL proxy variants (LD r2 ≥ 0.8) overlapped the affected caPeak and no other caPeak. We also tested driver peak models adjusting for the absolute value of caQTL effect size.
Stratified LD score regression
We used stratified LD score regression (Finucane et al. 2015) implemented in the LDSC software to test if liver caPeaks were enriched for heritability of specific traits. We downloaded GWAS summary statistics for 703 UKBB traits with a heritability significance of z ≥ 4 generated by the Benjamin Neale laboratory (https://nealelab.github.io/UKBB_ldsc/downloads.html). We converted the coordinates of caPeaks from GRCh38 to GRCh37 using liftOver (Hinrichs et al. 2006) and ran LDSC with the GRCh37 reference files provided with LDSC, which were built from 1000G phase 3 European genotypes. We restricted analyses to HapMap SNPs as previously described (Finucane et al. 2015). For each trait, we ran a model using liver caPeaks and the baseline v1.2 model. We considered two measures of significance: the P-value of the coefficient, which measures the contribution to heritability specific to liver caPeaks after removing contributions from all data sets in the baseline, and the P-value of the enrichment FC (Enrichment_p), which includes contributions to heritability from liver caPeaks that are shared with the baseline (Hormozdiari et al. 2018). We calculated coefficient P-values from the coefficient z-scores using a one-sided test assuming a standard normal distribution. We calculated FDR separately for enrichment P-values and coefficient P-values using the BH procedure (Benjamini and Hochberg 1995) and considered traits with FDR < 5% as significantly enriched.
Colocalization of GWAS signals with caQTLs and eQTLs
We downloaded GWAS summary statistics and signal lead variants for 17 traits: three liver enzyme traits (Pazoki et al. 2021), four lipid/cholesterol traits (Graham et al. 2021), four glycemic traits (Chen et al. 2021), type 2 diabetes (Mahajan et al. 2018), BMI (Yengo et al. 2018), waist–hip ratio adjusted for BMI (Pulit et al. 2019), vitamin D (Revez et al. 2020), coronary artery disease (Aragam et al. 2022), and CRP (Said et al. 2022). Although some of these GWAS studies consisted of multiple populations, we used lead variants and summary statistics from individuals genetically similar to European populations for all GWAS studies. We used conditionally distinct signal lead variants if they were provided by the study. For the two studies that did not report conditionally distinct signals (Graham et al. 2021; Said et al. 2022), we identified conditionally distinct lead variants using the following approach. We first defined loci from the GWAS marginal summary statistics by selecting the most significant variant with P < 5 × 10−8 in a region and removed all other variants within 500 kb (see Supplemental Methods). Loci whose leads were within 1 Mb of each other were merged. We then identified conditionally distinct lead variants within each locus using GCTA (Yang et al. 2011) cojo-slct using variants with MAF ≥ 1%, a collinearity threshold of 0.5, and LD estimated from about 40,000 UKBB donors genetically similar to European populations and extending 500 kb on either side of the lead variant(s) for the locus. For all studies, we performed colocalizations with conditional summary statistics generated using GCTA (Yang et al. 2011) cojo-cond for all signals in which the lead variant was within 500 kb of the lead variant of another signal; we performed colocalizations using marginal summary statistics otherwise. We used liftOver (Hinrichs et al. 2006) to convert coordinates of GWAS variants to GRCh38 to match the caQTL and eQTL data. For caQTLs, we used the summary statistics from all donors. For eQTLs, we used the summary statistics from donors similar to European populations and restricted to protein-coding and lncRNA genes. We tested for colocalization between a GWAS signal and either a caQTL or eQTL signal using coloc.abf (Giambartolomei et al. 2014) if the lead variants of the signals had LD r2 ≥ 0.5 (TOPMed Europeans). We ran coloc using betas and standard errors, and only used variants with MAF ≥ 1%. We considered signals that had posterior probability of colocalization (PP H4) > 0.8 to be colocalized. We used LocusZoom v1.4 (Pruim et al. 2010) to make plots of colocalized signals.
Because GWAS signals may be shared across traits, we estimated the number of unique colocalized GWAS signals across traits by first LD-clumping (r2 > 0.5, 1000G Europeans) (The 1000 Genomes Project Consortium 2015) GWAS leads across all traits and then counting the number of clumped signals that had at least one variant colocalized with a caQTL. We used the r2 > 0.5 threshold to avoid overcounting signals.
Prioritizing caQTLs signals for functional follow-up
We considered caQTLs for functional follow-up if they colocalized with an eQTL and a GWAS signal for lipid or liver enzyme traits and if the caPeak was in a set of coordinately regulated peaks. Because our CRISPRi experiments were performed in HepG2, we additionally required that the caPeak overlap an HepG2 ATAC peak (ENCODE accession ENCSR291GJU) (The ENCODE Project Consortium et al. 2020), that the predicted target gene from the eQTL was expressed in HepG2 (ENCODE accession ENCSR181ZGR), and that the genotype of the caQTL lead variant in HepG2 (ENCODE accession ENCFF989GDN) was either heterozygous or homozygous for the more accessible allele (Zhou et al. 2019).
Transcriptional reporter luciferase assays
We tested for allelic differences in transcriptional activity in HepG2 cells (López-Terrada et al. 2009) for variants overlapping caPeaks at the TENM2 and RALGPS2 loci. HepG2 cells were cultured in MEM-alpha supplemented with 10% FBS and 1 mM sodium pyruvate and maintained at 37°C with 5% CO2. We designed PCR primers to amplify DNA elements spanning predicted regulatory variants within caPeaks (Supplemental Table 25). For RALGPS2, we generated PCR products for both alleles using DNA from individuals homozygous or heterozygous for the variants, and for TENM2, we also used site-directed mutagenesis (Agilent QuikChange) to generate products with the alternate allele and to make other variable sites identical. We cloned the products into luciferase reporter vector pGL4.23 (Promega) as previously described (Fogarty et al. 2014). The day before transfection, we plated 90,000–120,000 cells into collagen-coated 24-well plates. We transfected 8 wells for each of four sequence-verified constructs for TENM2, and duplicate wells with eight sequence-verified constructs for RALGPS2. We cotransfected wells with phRL-TK Renilla reporter vector using lipofectamine 3000 (Life Technologies). We measured firefly luciferase activity 48 h after transfection and normalized to Renilla activity. We calculated the FC in luciferase activity relative to an empty pGL4.23 vector and used two-tailed t-tests to detect allelic differences in activity. We repeated experiments on a separate day and obtained similar results.
Selection and cloning of gRNA lentiviruses
We designed 10 forward and reverse gRNAs to target a 430 bp region (Chr 1: 178,551,259–178,551,689) within caPeak21014 using CRISPOR (Supplemental Table 26; http://crispor.tefor.net/). We used nontargeting control (NTC) gRNAs as described previously (Smith et al. 2022). We cloned all gRNAs as a pool into the Lenti U6-sgRNA/EF1a-mCherry vector (a gift from Jeremy Day, Addgene 114199) using BbsI-HF and a modified cloning protocol (Ran et al. 2013). Briefly, we annealed 10 µM forward and reverse gRNA primer pairs, phosphorylated using T4 PNK and T4 ligase buffer (NEB), and pooled the 10 gRNA blocks. We ligated 30 ng of BbsSI-HF-digested and calf intestinal phosphatase (CIP)–treated vector with 0.05 µM of annealed and phosphorylated gRNA oligos using T4-DNA ligase (NEB) for 4 h at 16°C, used 2 µL of the ligated product to transform 25 µL of stable competent cells (NEB), plated 125 µL of transformed cells onto ampicillin agar plates, gently scraped all more than 1000 colonies into 250 mL LB broth with ampicillin, and performed overnignt shaking at 37°C. We prepared plasmid DNA (HiPure plasmid maxiprep kit, Invitrogen) and confirmed the presence of all gRNAs by colony PCR.
We produced gRNA lentiviruses as described previously (Perrin et al. 2021). Briefly, we grew 4 × 106 HEK293T cells on a 10 cm plate; cotransfected with 9.5 µg lenti U6-sgRNA/EF1a-mCherry construct, 8 µg of packaging plasmid (psPAX2, a gift from Didier Trono, Addgene plasmid 12260), and 2.5 µg of an envelope plasmid (pMD2.G, a gift from Didier Trono, Addgene plasmid 12259) using Lipofectamine 2000 and PLUS reagent (Invitrogen); and replaced growth media after 18 h. We harvested viral supernatants at 48 and 72 h after transfection and concentrated using Lenti-X concentrators (Clontech). We functionally tittered the lentivirus by amplifying viral DNA WPRE, plasmid backbone DNA, and genomic DNA (LP34) for each sample as previously described (Gordon et al. 2020) and represented functional titers as multiplicity of infection (MOI).
CRISPRi
We used a doxycycline-inducible CRISPRi system (López-Terrada et al. 2009) by stably expressing dCas9 fused to a KRAB-repressor domain in the AAVS1 safe-harbor locus (Hazelbaker et al. 2020) as previously described (Pandey et al. 2024). We induced HepG2-dCas9-KRAB cells with doxycycline (2 µg/mL) for 48 h, sorted for GFP using a Becton Dickinson FACSAria II at the UNC Flow Cytometry Core Facility, and plated 1.2 × 105 GFP-positive cells/well onto collagen-I (Corning)-coated 12-well plates. We maintained cells in 275 µg/mL neomycin except during the transduction period. We spin-infected cells with lenti-gRNAs (Perrin et al. 2021) at 40 MOI in the presence of 10 µg/mL polybrene. After 8 h, we replaced growth media with media containing doxycycline (2 µg/mL) and neomycin and visualized GFP and mCherry expression by fluorescent microscopy. Ninety-six hours after transduction, we lysed cells, extracted total RNA (RNeasy plus, Qiagen), and converted to cDNA (Superscript IV Vilo, Invitrogen). We assessed gene expression by quantitative PCR using TaqMan probes for RALGPS2 (Thermo Fisher Scientific) and normalized to B2M (Hs00187842-m1) expression level. We performed experiments on two separate days and obtained similar results. For the second experiment, we removed one replicate from the nontargeting control experiments that was more than two standard deviations from the mean of gene expression.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE264684. Supplemental Data files containing ATAC peak locations, summary statistics for caQTL data, and motifs disrupted by caQTLs are available in the Supplemental Material, at Zenodo (https://zenodo.org/records/15025748), and at https://mohlke.web.unc.edu/data/. Scripts are provided as Supplemental Code.
Competing interest statement
F.I. is an employee of BeiGene and received stocks from the company. He also owns stocks of AbbVie.
Acknowledgments
This study was supported by National Institutes of Health (NIH) grants R01DK072193 and UM1DK126185. Individuals were supported by the American Heart Association POST903990 (G.K.P.) and NIH grants T32HL069768 (H.J.P.) and T32HL129982 (J.D.R.). We thank the donors of liver samples, which were obtained from the Liver Tissue Cell Distribution System supported by NIH contract N01DK70004/HHSN267200700004C. We thank Erika Deoudes for graphic design services. We thank the Roadmap Epigenomics Project for liver tissue chromatin states; the ENCODE Consortium for liver ATAC-seq, RNA-seq, and transcription factor ChIP-seq data; Cis-BP for TF binding motifs; and GTEx for liver tissue gene expression data. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. The data used for the analyses described in this paper were obtained from the GTEx Portal.
Author contributions: K.W.C., H.J.P., and K.L.M. designed the study. K.W.C. and A.A.A. performed bioinformatic analyses. H.J.P., S.V., and A.S.E. performed sample handling and ATAC-seq. G.K.P., S.V., and A.E.M. performed functional assays. A.S.C., E.G.S., and F.I. provided liver tissue samples. A.S.E., P.J.G., F.A.W., and Y.Z. provided access to resources. A.S.E., K.A.B., J.D.R., A.V., S.C.J.P., and L.M.R. provided analysis support. K.W.C. and K.L.M. wrote the manuscript. All authors reviewed and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279741.124.
- Received July 1, 2024.
- Accepted May 15, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








