
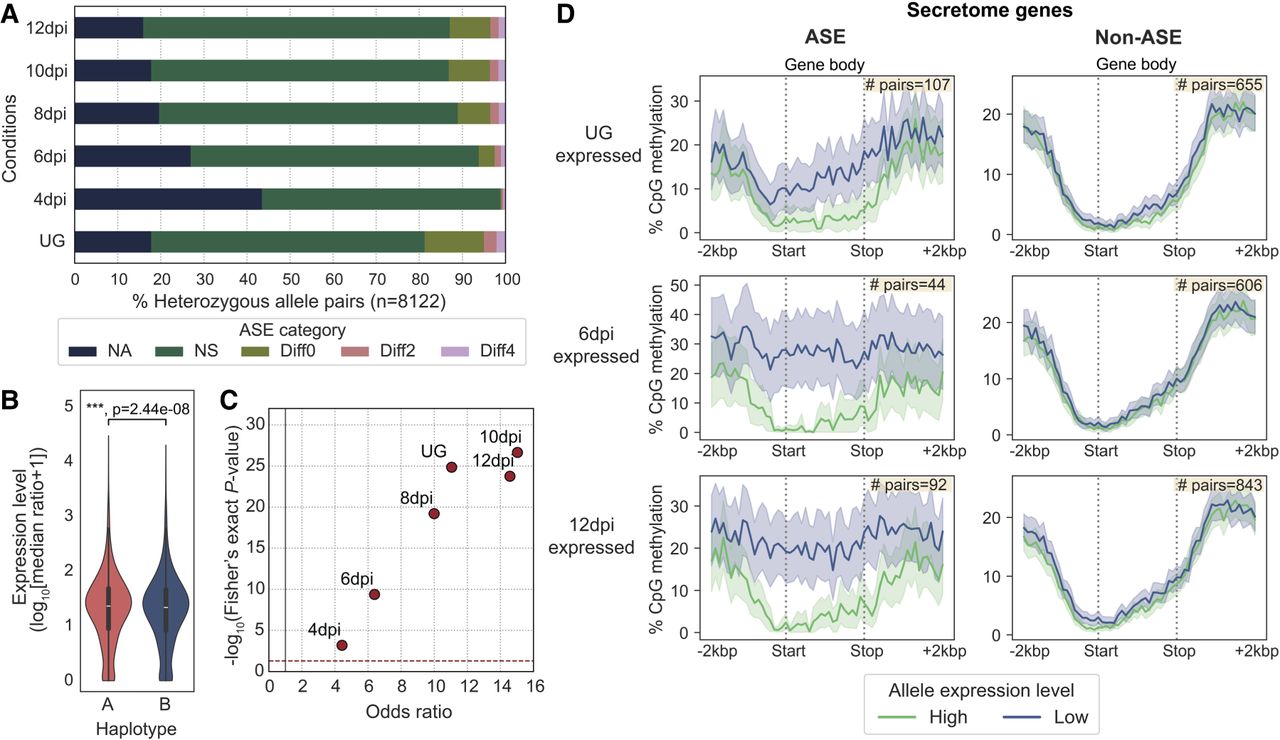
ASE is prevalent among secretome genes and correlates with gene methylation patterns. (A) The five ASE categories detected across six different transcript sampling conditions. Allele pairs displaying absolute log2 fold changes greater than two (Diff2 and Diff4) in at least one condition were defined as ASE in subsequent analysis. (B) Expression levels of alleles belonging to the two nuclear haplotypes A and B. Mann–Whitney U test: (***) P < 0.001. (C) Odds ratios and log10 P-values from Fisher's exact tests comparing the proportion of ASE genes among secretome genes and evolutionarily conserved BUSCOs. Each red dot represents a transcript sampling conditions as labeled. Dotted red and gray lines highlight cut-offs matching the null hypothesis of no difference between the two gene groups. (D) Distribution of the percentage of CpG methylation (sampled at UG) across ASE or non-ASE secretome gene bodies (here defined as start to stop codon) including ±2 kbp flanking regions. Solid lines represent mean percentage of CpG methylation; shaded areas represent 95% bootstrapping confidence intervals. Yellow inset shows the number of allele pairs included.








