
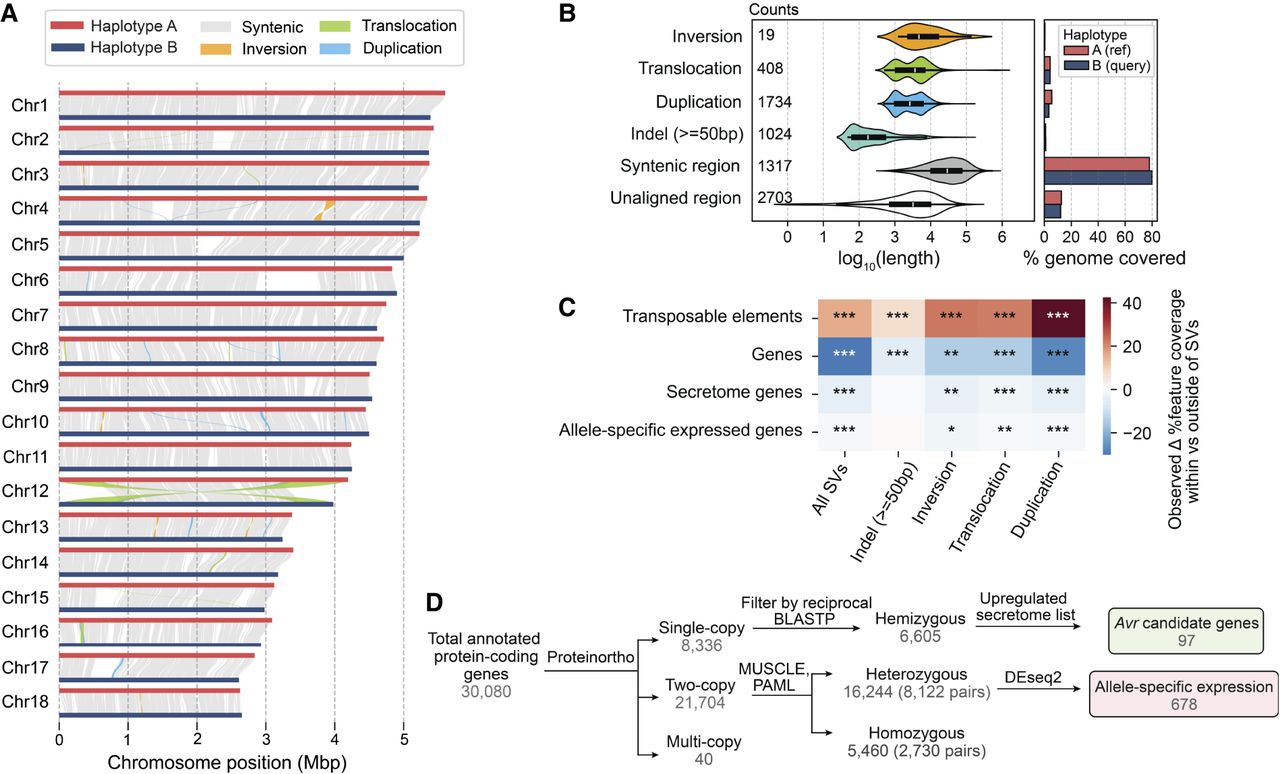
Pst104E’s inter-haplotype variation is shaped by large-scale structural variation (SV) and TEs. (A) Synteny and structural rearrangements between chromosome pairs of the two nuclear haplotypes, with A as reference and B as query. (B) Length distribution (left) of different SV types, including syntenic and unaligned regions. Counts of each SV type are shown. The bar chart (right) represents the total genome length covered by each sequence category. (C) Enrichment (red) and depletion (blue) of different genomic features (TEs and genes) within SVs including ±2 kbp flanking regions tested using permutations. The color scale denotes the observed difference in the percentage of coverage of a genomic feature located within and outside of a given set of SVs. P-values indicate the proportion of permuted coverage values less than, equal to, or greater than the observed: (*) P < 0.05, (**) P<0.01, (***) P<0.001, FDR-corrected. (D) Flowchart summarizing the classification of hemizygous, heterozygous, and homozygous protein-coding genes and their further categorization. Hemizygous genes were intersected with secretome genes upregulated during infection time points (4, 6, or 8 dpi) to shortlist Avr candidates. Heterozygous biallelic genes were used for allele-specific expression (ASE) analysis.








