
Cohesin organizes 3D DNA contacts surrounding active enhancers in C. elegans
Abstract
In mammals, cohesin and CTCF organize the 3D genome into topologically associating domains (TADs) to regulate communication between cis-regulatory elements. Many organisms, including S. cerevisiae, C. elegans, and A. thaliana contain cohesin but lack CTCF. Here, we used C. elegans to investigate the function of cohesin in 3D genome organization in the absence of CTCF. Using Hi-C data, we observe cohesin-dependent features called “fountains,” which have also been reported in zebrafish and mice. These are population average reflections of DNA loops originating from distinct genomic regions and are ∼20–40 kb in C. elegans. Hi-C analysis upon cohesin and WAPL-1 depletion supports the idea that cohesin is preferentially loaded at sites bound by the C. elegans ortholog of NIPBL and loop extrudes in an effectively two-sided manner. ChIP-seq analyses show that cohesin translocation along the fountain trajectory depends on a fully intact complex and is extended upon WAPL-1 depletion. Hi-C contact patterns at individual fountains suggest that cohesin processivity is unequal on each side, possibly owing to collision with cohesin loaded from surrounding sites. The putative cohesin loading sites are closest to active enhancers, and fountain strength is associated with transcription. Compared with mammals, the average processivity of C. elegans cohesin is about 10-fold shorter, and the binding of NIPBL ortholog does not depend on cohesin. We propose that preferential loading and loop extrusion by cohesin is an evolutionarily conserved mechanism that regulates the 3D interactions of enhancers in animal genomes.
In mammalian genomes, cohesin and CTCF form topologically associating domains (TADs) (da Costa-Nunes and Noordermeer 2023). The earlier models of TAD formation encompassed three main features: Cohesin is uniformly loaded; cohesin performs two-sided loop extrusion; and loop extrusion is stalled at CTCF-binding sites located at TAD boundaries (Fudenberg et al. 2017). The second and the third assumptions of the model were visualized in vitro using single-molecule assays, thus bridging the gap between the theoretical model and the experimental observation in vivo (Davidson et al. 2019; Kim et al. 2019). Further studies refined the earlier models and proposed mechanistic variations of two-sided loop extrusion (e.g., frequent switching of direction in a one-sided extrusion scheme), effect of cohesin molecules meeting one another (resulting in bypassing to form secondary folds in loops or colliding and stopping extrusion on one side), and specific proteins controlling loop extrusion as loader/facilitator (NIPBL) or remover (WAPL-1) or acting as barriers (e.g., MCM complex, RNA Pol II [Pol II], DNA topology) (Dequeker et al. 2022; Morao et al. 2022; Banigan et al. 2023; Corsi et al. 2023).
Recent studies also required an update to the uniform loading assumption, because Hi-C-detected features termed “jets,” “plumes,” or “fountains” depicted as secondary diagonals protruding from narrow segments suggest cohesin to be loaded at specific sites (Wike et al. 2021; Guo et al. 2022; Shao et al. 2024). The preferential loading sites locally increase the frequency of cohesin-mediated loops, thus providing another layer of 3D genome folding with the potential to regulate contacts between cis-regulatory elements. Such a mechanism of genome folding could have strong regulatory implications for species that contain tissue-specific enhancers but lack CTCF, such as Caenorhabditis elegans.
C. elegans is a small nematode with an ∼100 Mb genome and ∼20,000 genes (Heger et al. 2009). Median gene length is ∼2 kb, and intergenic distances between genes (excluding operons encompassing ∼15% of genes) range from ∼2 to 10 kb (Nelson et al. 2004; Girard et al. 2007; Allen et al. 2011). Like other species, enhancers are distinguished from promoters based on enrichment of distinct and conserved histone modifications, including H3K27 acetylation and H3K4 methylation (Evans et al. 2016; Daugherty et al. 2017). Mapping enhancer chromatin features and analyses of individual genes suggest that most enhancers are located ∼2 kb from promoters, but some enhancers can be up to ∼12 kb away (Shi et al. 2009).
In humans, somatic cohesin is composed of four subunits, SMC1, SMC3, RAD21, and STAG (Hassler et al. 2018). The STAG subunit has two isoforms, STAG1 and STAG2, that form cohesin in a mutually exclusive manner (Alonso-Gil et al. 2023). Several subunits of mammalian cohesin are specific for meiosis (Ishiguro 2019). In C. elegans, three of the four subunits of cohesin, SMC-1, SMC-3, and SCC-3 (STAG), have a single copy, whereas the kleisin subunit has five variants: COH-1, SCC-1 (also known as COH-2), COH-3, COH-4, and REC-8 (Wood et al. 2010; Severson and Meyer 2014). COH-2, but not COH-1, has mitotic functions, whereas COH-3/4 (paralogs) and REC-8 have meiotic functions (Mito et al. 2003; Severson and Meyer 2014; Hernandez et al. 2018; Yu et al. 2023). Here, we asked how cohesin contributes to 3D organization of the C. elegans genome.
Results
Fountains are a feature of 3D genome organization detected by Hi-C
C. elegans genome is organized into six similar-sized chromosomes, including five autosomes (I–V) and one X Chromosome. The chromosomes are holocentric and are partitioned into three. The gene-rich central regions make inter-chromosomal contacts, and gene-poor left and right arms are frequently located close to the nuclear lamina (Liu et al. 2011; Ho et al. 2014; Bian et al. 2020). At the megabase scale, the autosomes display A/B compartmentalization without loop-anchored TADs, which is consistent with the lack of CTCF (Fig. 1). The X Chromosomes contain weaker compartments along with loop-anchored TADs, formed by a condensin I variant that functions in dosage compensation (DC) in hermaphrodites (Crane et al. 2015; Anderson et al. 2019; Bian et al. 2020; Kim et al. 2022; Morao et al. 2022; Sawh and Mango 2022). At the multikilobase scale, we observe contact enrichment patterns that orthogonally protrude from the main diagonal across all chromosomes (Fig. 1).
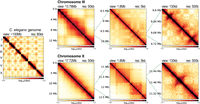
3D organization of the C. elegans genome at different scales. Leftmost panel shows the Hi-C contact matrix of the entire genome. The right three columns show an example of an autosome (top) and X Chromosome (bottom) at different scales. At the chromosome-wide scale (first column), autosomes show clear separation between the two interacting flanking arms and the center, whereas the X Chromosome is more uniform. At the megabase scale (second column), autosomes show checkerboard pattern, whereas X Chromosome additionally harbors TADs. At the kilobase scale (last column), both autosomes and X Chromosome show fountains that appear as protruding 3D DNA contacts from the main diagonal.
DNA loops originating and extending symmetrically in both directions from a specific site in a population of cells form enriched Hi-C contacts perpendicular to the main diagonal. Such patterns were termed “chromatin jets” in mice (Guo et al. 2022). Recent studies expanded on similar Hi-C features and defined “fountains,” which are not perfectly perpendicular and show “fanning,” owing to heterogeneity of the formed loops (Galitsyna et al. 2023; Isiaka et al. 2023; Shao et al. 2024). In the absence of quantitative criteria that distinguish chromatin jets and fountains, in our C. elegans Hi-C data we will refer to the contacts protruding from the main diagonal as “fountains” to be more inclusive of fanning.
To test whether the fountains are owing to loop extrusion by cohesin, we performed Hi-C analysis in the absence of cohesin or WAPL-1. Although cohesin depletion is sufficient to test for requirement, we reason that WAPL-1 depletion is necessary to distinguish loop extrusion from other mechanisms. For instance, if the fountains were formed by cohesin without a gradual increase in the DNA loop size, increasing the residence time of cohesin would result in more pronounced fountains of the same size. In contrast, the loop-extrusion model predicts that WAPL-1 depletion leads to cohesin translocating farther, extending the DNA loops and, thus, fountains in length.
Acute depletion of SMC-3 and WAPL-1 from somatic cells
In C. elegans, there is a singular subunit for SMC-1, SMC-3, and SCC-3 (Fig. 2A). To study the effect of cohesin loss, we targeted SMC-3 subunit for auxin-inducible degradation. WAPL-1 is encoded by wapl-1 in C. elegans (Gandhi et al. 2006; Kueng et al. 2006). The two proteins SMC-3 and WAPL-1 were C-terminally tagged with degron-GFP at the endogenous locus for auxin-inducible depletion, as employed previously (Zhang et al. 2015; Morao et al. 2022). To enrich postmitotic interphase cells, we used the early L3 stage, when most somatic cells have stopped dividing and the germline has not significantly proliferated (Sulston and Horvitz 1977). Furthermore, we depleted the proteins only in somatic cells, using a strain in which the auxin-response gene TIR1 is under the control of a soma-specific promoter eft-3 (Zhang et al. 2015). This background strain is used as a control, in which TIR1 is expressed, but none of the endogenous genes are tagged with degron-GFP (Fig. 2B).
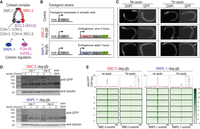
Acute depletion of cohesin and WAPL-1 from somatic cells. (A) C. elegans subunits of cohesin and its regulators. (B) Transgenic strains used in this paper. Exogenously inserted TIR1 is expressed in somatic cells using the eft-3 promoter. Endogenous smc-3 or wapl-1 are C-terminally tagged with degron-GFP. (C) DAPI and GFP images of L3 stage worms. Arrow indicates that the proteins in the germline are not degraded upon auxin treatment owing to TIR1 presence only in the somatic cells. Scale bar, 100 μm. (D) Western blot images of L3 stage worms with increasing auxin treatment duration. Anti-GFP antibody was used to show global depletion of the tagged protein. Antitubulin antibody was used as loading control. Asterisk indicates background bands. (E) Metaplot of ChIP-seq data grouped into different chromosomes. Anti-GFP antibody was used to show uniform depletion of SMC-3 and WAPL-1.
Upon visualizing DAPI and GFP, we observe that in the control condition, both cohesin and WAPL-1 show ubiquitous GFP signals, suggesting that cohesin and WAPL-1 are present in the interphase, as well as the few germ cells present in early L3 larvae (Fig. 2C, no auxin). Upon auxin treatment, SMC-3 and WAPL-1 are noticeably depleted in somatic cells but are maintained in the germ cells as expected from somatic expression of TIR1 (Fig. 2C, 1 h auxin). At population average, western blot analysis of degron-GFP-tagged proteins show noticeable depletion in the total amount of protein (Fig. 2D). This depletion is maximally achieved by 1 h treatment, and no further depletion was observed upon 2 h treatment. Therefore, we chose 1 h as the “depletion condition.” Lastly, we performed ChIP-seq using anti-GFP and observed depletion across the genome (Fig. 2E). In summary, both SMC-3 and WAPL-1 are acutely depleted from somatic cells with 1 h auxin treatment.
DNA loops forming the fountains are cohesin dependent and extend upon WAPL-1 depletion
Fountains are heterogeneous. They can be left-tilted (Fig. 3A) or right-tilted (Fig. 3B) or can show complex trajectories (Supplemental Fig. S1A). In all cases, SMC-3 depletion eliminated, and WAPL-1 depletion extended fountains. For example, in the control condition, we observed a short fountain (Fig. 3A, left). In the SMC-3 depletion, the fountain is lost, indicating that it is cohesin dependent (Fig. 3A, center). In the WAPL-1 depletion, the trajectory of the fountain is extended, suggesting that cohesin initially forms a smaller loop and then a larger loop with increasing residence time on DNA (Fig. 3A, right). The tilt of the fountains indicates that the processivity of cohesin is not equal on each side (Fig. 3C).
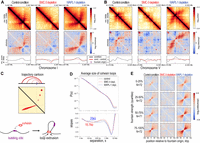
Cohesin forms fountains via loop extrusion. (A) Example Hi-C snapshot of left-directed fountain in control, SMC-3-, and WAPL-1-depletion conditions. (From top to bottom rows) The observed balanced matrix, distance-decay normalized observed-over-expected matrix, and insulation score using 30 kb window and 500 bp step. Black lines indicate the condition (control of depletions); red lines, the condition minus the control; and the purple vertical line, identified fountain origins. (B) Example Hi-C snapshot of right-directed fountain. (C) Cartoon trajectory of left-directed fountain (A) arising from two-sided loop-extruding factor reeling in DNA faster on the left than right side. (D) Contact probability, P(s), as a function of linear genomic separation between pairs of loci, s (top), and its log-derivative, the slopes of P(s) in log-space (bottom), are plotted for three conditions in different colors. The inferred average loop size for cohesin in unperturbed state (red) and the extended average loop size for cohesin in WAPL-1 depletion (blue) are annotated. (E) On-diagonal pileup of observed-over-expected matrix centered at the fountain origins grouped into four quantiles based on the fountain strength.
In vitro, cohesin performs two-sided loop extrusion (Davidson et al. 2019; Kim et al. 2019). In simulation, cohesin was inferred to be an effectively two-sided loop-extruding factor (Banigan et al. 2020). Hi-C data cannot distinguish if cohesin acts as a single molecule pulling DNA at both ends or two tethered molecules each extruding from one end. Regardless, our observation that cohesin mediates DNA contacts protruding from the Hi-C diagonal supports the conclusion that cohesin effectively functions as a two-sided loop-extruding factor in vivo. Furthermore, detection of cohesin loops in the Hi-C assay supports the idea that they are formed by preferential cohesin loading at stereotypical locations.
Cohesin-mediated DNA loops in C. elegans are shorter than those observed in mammals
Although fountains are pronounced Hi-C features, they may reflect above-average cohesin behaviors on chromatin. The contact probability curve, P(s), and its log-derivative are powerful metrics for estimating the average loop size of the chromosome (Gassler et al. 2017; Polovnikov et al. 2023). In the mammals, this approach revealed that cohesin forms ∼100–200 kb loops on average (Haarhuis et al. 2017; Davidson et al. 2019; Polovnikov et al. 2023). Here, we use a similar metric to analyze the size of loops formed by cohesin. Although a local maximum is present between the 100 kb and 1 Mb range, this does not correspond to cohesin loops, given it does not change in SMC-3- or WAPL-1-depletion conditions and is larger than observed fountains. Instead, a local maximum appears in WAPL-1 depletion at ∼25 kb on autosomes (Fig. 3D; Supplemental Fig. S1B). We infer that the average size of DNA loops mediated by cohesin in the control condition is smaller and may correspond to local minima that becomes more pronounced in SMC-3 depletion, which is near ∼15 kb on autosomes. The cohesin-mediated DNA loops are shorter on the X compared with autosomes (Supplemental Fig. S1B). This may be related to the activity of a DC-specific condensin that forms TADs and represses transcription (Albritton and Ercan 2018; Kim et al. 2022). We conclude that the loops formed by C. elegans cohesin are about 10-fold smaller than the ones formed by the mammalian cohesin.
Cohesin binding tracks the trajectory of the fountains and extends farther upon WAPL-1 depletion
Our Hi-C analysis is consistent with a model of cohesin preferentially loading and loop extruding to form fountains. An orthogonal approach to test if the observed fountains are formed by the loop-extruding cohesin is to analyze the distribution of cohesin with respect to the fountain origins. To identify fountains genome-wide, we determined sites across which DNA contacts are facilitated by cohesin. By our definition, cohesin-dependent fountains must be present in both the control and WAPL-1-depletion conditions and show significant decrease upon SMC-3 depletion (Supplemental Fig. S1C). A total of 287 sites satisfied the criteria. Meta-analysis of the average Hi-C signal in quartiles of “fountain strength” (see Methods) confirmed the identification of the origins of cohesin-dependent fountains (Fig. 3E) and was consistent between experimental replicates (Supplemental Fig. S1D).
In simulation, loop-extruding factors with a preferential loading site generate a “spreading” pattern, in which binding gradually decreases with increasing genomic distance from a loading source (Brandao et al. 2021). Loop extruders with known recruitment mechanisms support this pattern. ChIP-seq enrichment of bacterial condensin spreads out from the parS sequences (Sullivan et al. 2009; Wang et al. 2017, 2018). Similarly, C. elegans condensin DC shows reduced binding with increasing distance from the endogenous or ectopically inserted recruitment elements on the X (rex sites) (Kim et al. 2022; Morao et al. 2022). Targeted loading of cohesin using DNA damage or the tetO-tetR system also shows spreading of cohesin to nearby regions (Arnould et al. 2021; Han et al. 2023). Therefore, if cohesin is preferentially loaded at fountain origins, we predict cohesin binding to show a spreading pattern. Furthermore, under WAPL-1 depletion, in which the fountain length increases, the spreading pattern of cohesin would widen as cohesin travels farther away.
To analyze cohesin binding, we performed ChIP-seq for two subunits of the cohesin complex, SMC-1 and SMC-3. In the control condition, we observe that cohesin binds broadly for the span of ∼20 kb region (Fig. 4A; Supplemental Fig. S2, black tracks). Upon SMC-3 depletion, both SMC-3 and SMC-1 are largely depleted (Fig. 4A, red tracks). For SMC-1, we observe residual binding at the fountain origins (Fig. 4B), which is consistent across biological replicates (Supplemental Fig. S3A) and is unlikely owing to differences in the specificities of SMC-3 and SMC-1 antibodies to the fountain origins (Supplemental Fig. S3B). The residual SMC-1 may reflect the process of searching through its potential target sites (Izeddin et al. 2014) or transient association as observed with the cohesin complex with defective ATPase activity (Ladurner et al. 2014). The loss of SMC-1 and SMC-3 in the flanking regions is consistent with the loss of the fountain in the Hi-C data (Fig. 3), demonstrating that cohesin is essential for fountain formation.
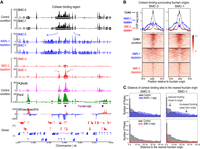
Cohesin spreads from fountain origins. (A) Genome browser view around fountain origins. Plotted are the input-subtracted ChIP-seq tracks for cohesin subunits, SMC-3 and SMC-1, in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). The ticks below the signal tracks indicate MACS2-called peaks. Fountain origins are shown in purple. (B) Average profile and heatmap of SMC-3 and SMC-1 with respect to fountain origins. Plotted are the input-subtracted ChIP-seq tracks for the cohesin subunits SMC-3 and SMC-1 in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). (C) Histogram of distance between cohesin subunit summit and the nearest fountain origin. The three conditions are control (gray), WAPL-1 depletion (blue), and SMC-3 depletion (red). Change in cohesin binding sites upon WAPL-1 depletion is consistent between SMC-1 and SMC-3 and is highlighted in the panel corresponding to SMC-1.
Upon WAPL-1 depletion, the cohesin binding region widens, reaching a span of ∼40 kb (Fig. 4A; Supplemental Fig. S2, blue tracks). The increase in cohesin spreading upon WAPL-1 depletion is reproducible (Supplemental Fig. S3A) and generalizable across the fountain origins as measured by intensity (Fig. 4B) or by binding site distance to the fountain origins (Fig. 4C). This demonstrates that depletion of WAPL-1 results in cohesin moving farther away from the fountain origins. If cohesins were moving toward the fountain origins, WAPL-1 depletion would increase cohesin binding at the fountain origin. In contrast, WAPL-1 depletion results in relatively lower cohesin at the fountain origins (Supplemental Fig. S3C). Thus, we conclude that cohesins that are loaded at the fountain origins are moving away from the loading site in both directions, consistent with preferential loading followed by bidirectional loop extrusion implied by the Hi-C data (Fig. 3C).
Fountain origins harbor multiple PQN-85 binding sites
NIPBL protein is important for cohesin recruitment to DNA (Ciosk et al. 2000). Thus, we next analyzed the distribution and function of the C. elegans ortholog of NIPBL, encoded by pqn-85/scc-2 (Lightfoot et al. 2011). The strength of the fountains positively correlated with both the average PQN-85 ChIP-seq signal (Fig. 5A, left) as well as the number of PQN-85 peaks (Fig. 5A, right), including those present in the majority of replicates (Supplemental Fig. S3D). Recent studies suggested that NIPBL is a processivity factor for cohesin loop extrusion (Ciosk et al. 2000; Kagey et al. 2010; Busslinger et al. 2017; Alonso-Gil et al. 2023; Banigan et al. 2023; Han et al. 2023). These models suggested that NIPBL is carried by cohesin to nonloading sites through loop-extrusion activity (Petela et al. 2018; Han et al. 2023). We tested this prediction for C. elegans by performing ChIP-seq for PQN-85 upon SMC-3 and WAPL-1 depletion.
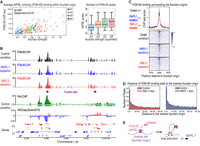
PQN-85 correlates with fountain strength, and its binding remains unchanged upon SMC-3 and WAPL-1 depletion. (A, left) Scatterplot of fountain strength versus mean PQN-85 ChIP-seq enrichment in control condition. Fountains divided into quartiles based on their strength are shown in different colors. (Right) Boxplot of fountain strength (divided into quantiles) and the number of PQN-85 summits within 6 kb region. (B) Genome browser view around fountain origins. Plotted are the input-subtracted ChIP-seq tracks for PQN-85 in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). The ticks below signal tracks indicate MACS2 called peaks. Fountain origins are shown in purple. (C) Average profile and heatmap of PQN-85 with respect to fountain origins. Plotted are the input-subtracted ChIP-seq tracks for cohesin subunits, SMC-3 and SMC-1, in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). (D) Histogram of the distance between PQN-85 summit and the nearest fountain origin. The three conditions are control (gray), WAPL-1 depletion (blue), and SMC-3 depletion (red). (E) Cartoon model of cohesin moving away from fountain origins without PQN-85 as it performs two-sided loop extrusion.
Cohesin and WAPL-1 depletion does not affect PQN-85 binding pattern measured by ChIP-seq
If cohesin mediates PQN-85 binding, a portion of PQN-85 ChIP-seq signal must be SMC-3 dependent. This prediction was not met. Unlike SMC-1, NIBPL binding surrounding the fountains did not reduce upon SMC-3 depletion (Fig. 5B,C, red tracks), and the position of PQN-85 peaks relative to the fountain origins did not change (Fig. 5D, left). The PQN-85 antibody was previously validated by western blot analysis upon immunoprecipitation and RNAi depletion (Kranz et al. 2013). To confirm that ChIP-seq signal is dependent on PQN-85 protein, we degron-tagged pqn-85 endogenously and validated knockdown by western blot analysis (Supplemental Fig. S4A,B). Binding of PQN-85 and SMC-3 surrounding the fountain origins was consistently lower upon PQN-85 depletion in the presence of auxin, whereas H4K20me1 did not change (Supplemental Fig. S4C–E). Therefore, PQN-85 ChIP-seq signal is dependent on PQN-85 protein, validating our conclusion that SMC-3 depletion did not significantly reduce PQN-85 binding at fountains.
A second prediction of PQN-85 translocating with cohesin is that upon WAPL-1 depletion, PQN-85 binding should also increase outward from the fountain origins. This prediction was also not met. Unlike SMC-3, the PQN-85 binding region did not become wider when WAPL-1 was depleted (Fig. 5B,C, blue tracks). We noticed variability in the level of WAPL-1 depletion between biological replicates and accounted for it by comparing PQN-85 and SMC-3 binding in the same ChIP extracts (Supplemental Fig. S5A). Although slightly more PQN-85 peaks were detected upon WAPL-1 depletion (Supplemental Fig. S5B), the effect was global and did not result in a relative increase in PQN-85 away from the fountain origins (Fig. 5D; Supplemental Fig. S5C) or a decrease at origins (Supplemental Fig. S5D). The lack of PQN-85 redistribution upon WAPL-1 depletion is supported both by calling peaks from individual replicates and by taking peaks called in majority of the replicates (Supplemental Fig. S5E).
Further categorizing cohesin binding sites into two groups, those uniquely present in the control condition and those in the WAPL-1-depletion condition, also shows that cohesin redistributes upon WAPL-1 depletion (Supplemental Fig. S6A) positioning farther away from the fountain origins (Supplemental Fig. S6B). Despite clear redistribution of cohesin, no such phenomenon was observed for PQN-85 (Supplemental Fig. S6C,D). Consistently, although cohesin binding decreased at the fountain origin upon WAPL-1 depletion (Supplemental Fig. S3C), PQN-85 binding did not (Supplemental Fig. S5D). Together, these results suggest that PQN-85 does not translocate significantly with cohesin in C. elegans (Fig. 5E).
Fountain patterns are influenced by the distribution of cohesin loading sites
If focal loading of cohesin is the basis of fountains, distribution of the NIPBL ortholog PQN-85 binding sites should correlate with the diversity of fountain patterns. Indeed, fountains with a broad base of interactions have a wide distribution of shorter PQN-85 ChIP-seq peaks near the fountain origin. For instance, a fountain with a broader PQN-85 binding region contains a small TAD-like feature that is converted to a fountain at a farther distance away from the main diagonal (Fig. 6A). The broad distribution of PQN-85 sites close to the origin of this fountain is consistent with simulation of “broad loading” producing TAD-like structures (Guo et al. 2022). Thus, we infer that the small TAD-like feature is caused by multiple cohesins colliding with each other, forming a randomly distributed cohesin stack. The extension into a fountain is likely caused by cohesins at the two ends of the stack that are not constrained (Fig. 6A, cartoon).
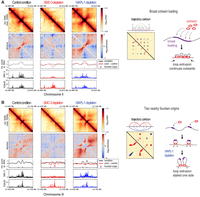
Fountains are heterogeneous. (A) Hi-C snapshot of a locus containing broadly distributed PQN-85 around fountain origins forming a TAD-like pattern. The trajectory cartoon shows one possible interpretation in which broadly loaded cohesins form multilayered nested loops (blue, green, purple), giving rise to a broad region of enriched contact. (B) Hi-C snapshot of a locus containing two nearby fountains forming “stripes.” The trajectory cartoon shows one possible interpretation, in which collision of two-sided loop-extruding cohesins from two fountain origins stall each other, causing them to behave as one-sided loop-extruding factors (red). The collision results in nested loops, in which the outer loop (blue) forms enriched contact away from the main diagonal.
Diversity of fountain patterns support collision and stalling of cohesin molecules in vivo
The increased cohesin processivity upon WAPL-1 depletion revealed possible collision between cohesin molecules. For example, two distinct fountains in the control condition extend to generate broad stripes upon WAPL-1 depletion (Fig. 6B). The stripes can emerge from cohesin coming from the left fountain origin stalled by the cohesin coming from the right fountain origin and vice versa. Alternatively, a bidirectional barrier between the two fountain origins stalls cohesins loaded at either fountain. For this particular region, we favor cohesin collision, because the pronounced contact probability that appears off-diagonal suggests the presence of a cohesin–cohesin nested structure (Fig. 6B, cartoon). This interpretation is supported by similar Hi-C patterns produced by simulation of two-sided loop-extruding factors from two loading sites with low bypass rate (Brandao et al. 2021).
Additional examples illustrate the heterogeneity of fountain patterns. A fountain with a simple trajectory shows a relatively narrow PQN-85 binding at the origin (Supplemental Fig. S7A), whereas another that contains multiple PQN-85 binding sites at the origin shows more complex Hi-C interactions and fountain trajectory (Supplemental Fig. S7B). In one example, we observe a mid-shift in the fountain's trajectory, suggesting that there is an uneven distribution of factors that regulate cohesin movement (Supplemental Fig. S7C). Lastly, we observe a “stripe-like” feature embedded within a fountain (Supplemental Fig. S7D), which could imply presence of a “stalling element” or a special case of loading event (Arnould et al. 2021; Han et al. 2023). We surmise that the diverse fountain shapes emerge from heterogenous PQN-85/loading site distribution and other in vivo barriers/facilitators of loop extrusion.
Fountain origins are close to active enhancers
To identify genomic elements that underlie the preferential loading of cohesins, we used genome annotations based on ChromHMM from two previous studies (Evans et al. 2016; Daugherty et al. 2017). Fountain origins are closest to genomic regions designated as active enhancers compared with other annotations (Fig. 7A; Supplemental Fig. S8A). Because the nearest enhancer–promoter pairs have a median distance of <2 kb in C. elegans (Supplemental Fig. S8B), fountain origins are often found in regions of high enhancer and promoter density (Supplemental Fig. S9). At a region where they are sufficiently distant, fountain origin was closer to the strong ATAC-seq peak associated with the enhancer (Fig. 7B; Supplemental Fig. S8C). Thus, preferential loading of cohesin forming the fountains occurs at sites close to active enhancers.
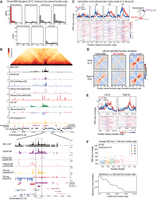
Fountain origins are close to active enhancers. (A) Histogram of genomic distance between identified fountain origins in this paper and the annotated regions of ChromHMM in (Daugherty et al. 2017). Active enhancers are highlighted in red. (B) Genome browser snapshot of Hi-C and other transcription related features such as topoisomerase-II, H3K27ac, H3K4me3, Pol II, GRO-seq, and ATAC-seq (top). Zoom-in near a fountain origin is shown at the bottom. (C) Average profile of GRO-seq data from Kruesi et al. (2013) split into plus (red) and minus (blue) strands is plotted with respect to the fountain origins. The fountains are grouped into four quantiles based on their strength. On the right, the cartoon highlights that origins of strong fountains are often close to upstream of transcription. (D) Left- and right-directed fountains were identified using the fontanka tool (Galitsyna et al. 2023). The fountain origins of the two groups are mutually exclusive. (E) GRO-seq plot centered around the left- and right-directed fontanka fountain origins. (F) Scatterplot showing relationship between fountain strength of the fountain origin and the signal mean of GRO-seq (Kruesi et al. 2013) in 20 kb region centered at the fountain origin (top). Sweep of Spearman's correlation as a function of flanking region used to average GRO-seq signal (bottom).
Plotting published GRO-seq data (Kruesi et al. 2013) suggests that fountain origins are located upstream of transcribed regions (Fig. 7C). To test if the direction of transcription correlates with trajectory of the fountains, we categorized them as left- and right-directed using the fontanka tool (Fig. 7D; Supplemental Fig. 10A–C; Galitsyna et al. 2023). Although there was some bias in the distribution of Pol II binding sites with respect to left- or right-tilted fountains (Supplemental Fig. S10D), correlation between the tilt and the direction of transcription was not strong (Fig. 7E). Nevertheless, three lines of analyses, including example fountains coinciding with histone modifications and proteins associated with active transcription (Fig. 7B; Supplemental Fig. S8C), GRO-seq enrichment being lower around weak fountains and higher around strong ones (Fig. 7C), and a positive correlation between fountain score and transcription surrounding a fountain origin (Fig. 7F), together demonstrate that fountains are associated with regions of active transcription.
One-hour depletion of SMC-3 or WAPL-1 does not lead to changes in RNA Pol II binding
We used ChIP-seq analyses of RNA Pol II to analyze the effect of SMC-3 and WAPL-1 knockdown. Unlike the striking changes observed in Hi-C, cohesin and WAPL-1 depletion did not result in significant change in RNA Pol II distribution (Fig. 8A). The lack of change in RNA Pol II ChIP-seq was reproducible (Supplemental Fig. S11A) and generalizable across fountain origins (Fig. 8B). Next, we categorized genes into two groups: one with higher cohesin binding in control and another upon WAPL-1 depletion (Fig. 8C). The noticeable shift in cohesin binding between the two groups of genes upon WAPL-1 depletion was not accompanied by changes in binding of RNA Pol II (Fig. 8D; Supplemental Fig. S6E). Orthogonally, mRNA-seq analyses showed minimal change upon 1 h depletion of SMC-3 and WAPL-1 (Fig. 8E). We considered the possibility that chromatin may require more time to reach a new-found equilibrium upon loop-extrusion perturbation (Nuebler et al. 2018). However, increasing the duration of auxin treatment to 4 h also showed minimal change in mRNA-seq (Supplemental Fig. S11B). Therefore, we conclude that acute depletion of cohesin and WAPL-1 change cohesin binding and the 3D DNA contacts measured by Hi-C but do not result in equally strong changes in gene expression.
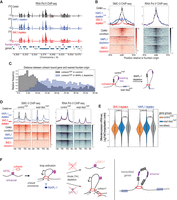
One-hour depletion of SMC-3 or WAPL-1 does not lead to drastic changes in RNA Pol II binding. (A) Genome browser view around fountain origins. Plotted are the input-subtracted ChIP-seq tracks for Pol II in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). The ticks below the signal tracks indicate MACS2-called peaks. Fountain origins are shown in purple. (B) Average profile and heatmap of SMC-3 and Pol II binding with respect to fountain origins. Plotted are the input-subtracted ChIP-seq tracks SMC-3 and Pol II in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). (C) Gene annotations from ce10 refGenes are categorized into cohesin high in control condition (gray) or in WAPL-1-depletion condition (blue), and their relative positions are plotted as a histogram from the nearest fountain origin. The two groups are mutually exclusive. The cartoon highlights that genes with higher cohesin binding in control are closer to fountain origins than genes with higher cohesin binding in WAPL-1-depletion condition. (D) Average profile and heatmap of SMC-3 and Pol II binding with over two groups of genes shown in C. Plotted are the input-subtracted ChIP-seq tracks SMC-3 and Pol II in three conditions: control (black), WAPL-1 depletion (blue), and SMC-3 depletion (red). (E) Differential mRNA-seq expression values (log2fc from DESeq2) are plotted for control high (n = 817), WAPL-1 high (n = 1268) genes that were further subsetted to be within 40 kb from the fountain origin, where the differential cohesin binding is the most pronounced. Other genes (n = 15,800) were used as control. Mann–Whitney U-test was used to generate P-values. (F) The proposed model of cohesin loading and loop-extrusion events inferred from the perturbation experiments and literature. Activated enhancers are bound by the NIPBL homolog PQN-85, which increases the frequency of cohesin loading at these sites. After loading, cohesin extrudes DNA in an effectively two-sided manner. Note that despite being depicted as a singular ring, the data cannot distinguish how many cohesin complexes are at the base of the loop. Upon acute depletion of SMC-3, the binding of PQN-85 is unaffected, and a residual amount of SMC-1 is found at loading site. The extruded loop is abolished. Upon acute depletion of the negative regulator WAPL-1, cohesin further extrudes DNA in two-sided manner away from the jet origin. The extended extrusion revealed that a clash between incoming and outgoing cohesin molecules may stall extrusion. Together, cohesin processivity at each side can be unequal and may be facilitated by transcription moving in the same direction. Despite the lack of immediate effect on transcription upon acute SMC-3 and WAPL-1 depletion, the extruded DNA loops encompass multiple cis-regulatory elements and active genes.
Discussion
Cohesin- and CTCF-mediated TAD formation contribute to proper development and normal physiology in mammals, but many organisms, including yeast, worms, and plants, contain cohesin but lack CTCF (Heger et al. 2012; Dong et al. 2017). Here, we used C. elegans as a model to address the following question: What does interphase cohesin do in multicellular organisms without CTCF? We reasoned that the answer would point to the ancestral function of cohesin in gene regulation. Our approach was to apply the auxin-inducible degradation system to acutely deplete the cohesin ATPase subunit SMC-3 or its negative regulator WAPL-1 in somatic cells in C. elegans larvae. Our results are consistent with a model that preferential loading of cohesin followed by bidirectional loop extrusion organizes the 3D DNA contacts surrounding active enhancers (Fig. 8F).
Fountains are a conserved feature of 3D genome organization mediated by cohesin
Recent studies, including this one, demonstrate that cohesin-mediated loops originating from distinct sites are the basis of Hi-C defined features termed jets, plumes, or fountains in model species of fungi, worms, zebrafish, and mammals (Liu et al. 2021a; Wike et al. 2021; Guo et al. 2022; Galitsyna et al. 2023; Isiaka et al. 2023; Shao et al. 2024). In zebrafish, fountains coincide with enhancer activity in zygotic genome activation, and knockdown of pioneer transcription factors eliminates fountains originating at these sites (Galitsyna et al. 2023). In mammalian cells, analysis of cohesin binding upon CTCF site deletion, depletion of pioneer transcription factors, and ectopic recruitment experiments suggest that cohesin is preferentially loaded at enhancers (Dowen et al. 2013; Liu et al. 2021b; Vos et al. 2021; Han et al. 2023). Our work also showed an overlap of fountain origins with active enhancers in C. elegans; thus, preferential loading of cohesin at enhancers may be an evolutionarily conserved mechanism of regulating their 3D DNA contacts.
NIPBL recruitment to active enhancers may facilitate cohesin loading
Based on analysis of individual fountains and their genomic context, we observed that the strength and the pattern of 3D DNA contacts forming the fountains correlate with the binding of the C. elegans NIPBL ortholog, PQN-85/SCC-2. Immunofluorescence analyses of cohesin in the germline showed that PQN-85 is required for cohesin binding to chromosomes (Ciosk et al. 2000; Seitan et al. 2006; Lightfoot et al. 2011). We also observed lower cohesin binding upon PQN-85 depletion (Supplemental Fig. S4). In many organisms, including C. elegans, NIPBL binds to active enhancers (Kranz et al. 2013; Busslinger et al. 2017; Zhu et al. 2021). In yeast, NIPBL interacts with and is recruited by the mediator complex (Mattingly et al. 2022). Therefore, it is possible that transcription factor interaction with the mediator recruits NIPBL to load cohesin at active enhancers.
In mammals, NIPBL is proposed to be a processivity factor that regulates the engagement of cohesin with DNA during loop extrusion (Alonso-Gil et al. 2023). ChIP-seq analysis of NIPBL upon RNAi depletion of a cohesin subunit in HeLa cells suggested that NIPBL binding at promoters is cohesin dependent (Banigan et al. 2023). Ectopic loading of cohesin by the tetO-tetR system showed that NIPBL binding spreads along with cohesin, suggesting that cohesin carries NIPBL as it moves (Han et al. 2023). In C. elegans, we did not observe a change in PQN-85 binding upon cohesin or WAPL-1 depletion (Fig. 5). Therefore, it is unlikely that a significant amount of PQN-85 protein translocates along with loop-extruding cohesin in C. elegans. It is possible that the longer processivity of mammalian cohesin compared with that of C. elegans is related to PQN-85's ability to translocate with cohesin in a larger genome. Differences between cohesin and its regulators in mammals versus C. elegans may provide insights into the conserved and distinct mechanisms during the evolution of animal genomes.
Collision of loop-extruding cohesins creating a barrier effect
Modeling approaches are used to connect the behavior of single cohesin molecules to the population average data produced in Hi-C, and we have interpreted our data in light of these studies (Banigan et al. 2020). Our results suggest that frequent loading results in the collision of loop-extruding cohesin molecules when these molecules are not removed from the DNA template by WAPL-1 (Fig. 6). The resulting structure suggests cohesin continues to loop extrude on one side while being stalled on the other. Therefore, an asymmetry in the loop extrusion is introduced by a barrier, in this case an incoming cohesin. Supporting our interpretation, computational modeling used the interaction of outgoing and incoming cohesin molecules to explain the dispersion property of fountains in zebrafish (Galitsyna et al. 2023), which are similar to the fountains in C. elegans and unlike the hairpin structures formed by preferential loading and extrusion by bacterial SMCs (Brandao et al. 2021). Furthermore, the loop-stacking model derived from in vivo imaging was proposed to emerge from collision among cohesins (Hafner et al. 2023).
Collision between loop extruders may be a general feature of SMC complexes. In C. elegans, a specialized condensin I complex for DC is loaded at distinct cis-regulatory elements on the X Chromosomes (Kim et al. 2022). The loading sites also form TAD boundaries by blocking loop extrusion by condensin DC (Anderson et al. 2019; Rowley et al. 2020). It is possible that the frequent loading of condensin DC at one recruitment site blocks the incoming molecules from another, because condensin DC loop extrudes in one direction (Kim et al. 2022). We observed that cohesin-mediated loops are shorter on the X Chromosome, raising the possibility that condensin DC may also stall cohesin; thus, different SMC complexes may also collide.
RNA Pol II: barrier or facilitator of cohesin-mediated loop formation?
Our cohesin ChIP-seq data in C. elegans produced a pattern consistently observed in many organisms, in which distinct peaks of binding are detected at promoters and enhancers (Peters et al. 2008; Rittenhouse and Dowen 2024). We also observe that prominent fountains are formed at regions of high transcription. Earlier studies in yeast suggested that RNA Pol II may push cohesin (Glynn et al. 2004; Lengronne et al. 2004). More recent work used the framework of loop-extrusion model and proposed that RNA Pol II and MCM complexes may be barriers to cohesin-mediated loop extrusion (Dequeker et al. 2022; Banigan et al. 2023). It is possible that transcription facilitates the movement of cohesins at fountains. Alternatively, intermittent stalling by polymerase allows for better capturing of the loops by cross-linking-dependent methods like Hi-C and ChIP-seq. Facilitation or barrier activity of transcription may be based on the transcription machinery itself or the associated changes to DNA topology (Morao et al. 2022; Guérin et al. 2024). Future work is needed to determine if the association between transcription and fountains is causal or correlational owing to sharing a common mechanistic origin.
Fountains and gene regulation
In our study, 1 h depletion of SMC-3 and WAPL-1 resulted in striking changes in cohesin binding and the Hi-C contacts forming fountains, but not on RNA Pol II binding or mRNA levels measured by ChIP-seq and mRNA-seq, respectively (Fig. 8). In a recent preprint, TEV-mediated cleavage of two cohesin kleisin subunits over a period of ∼24 h from L1 larvae to L3 was used to demonstrate the role of COH-1 in fountain formation and to address cohesin function in gene regulation in C. elegans (Isiaka et al. 2023). The investigators reported a slight increase in the expression of genes closer to fountain origins upon cohesin cleavage. This contrasts with the slight decrease we observed (Fig. 8E). In both studies, mRNA-seq was performed in whole animals, and the effect was subtle. Thus, further research is required to address cohesin function in gene regulation.
In mammals, the acute knockdown of cohesin also results in subtle changes in gene expression, yet mutations that reduce cohesin dosage or TAD boundaries produce significant changes (Cummings and Rowley 2022; Hafner and Boettiger 2023). It is possible that cohesin acts as a fine-tuner of transcription and/or functions earlier in development, and once an epigenetic state is established, the short-term depletion of cohesin does not result in changes in transcription (Pelham-Webb et al. 2020).
Evolution of cohesin activity
The coevolution of underlying linear genome complexity and cohesin function may explain CTCF being lost in C. elegans. In mammals, the relatively large cohesin loops are constrained by positioning of CTCF to prevent ectopic activation of promoters (Levine et al. 2014; Hnisz et al. 2016). C. elegans cohesin makes approximately 10-fold smaller loops compared with the mammalian cohesins, and this scales with corresponding gene density (about one gene per 5 kb vs. 100 kb). Our study offers two ideas for the shorter processivity of C. elegans cohesin. First in a compact genome, frequent encounters between incoming and outgoing cohesin molecules, as well as other SMC complexes and in vivo barriers, may limit cohesin translocation. Second, intrinsic factors may also reduce processivity. For instance, NIPBL may have a more limited role in the processivity of cohesin in C. elegans compared with that in mammals.
Whether cohesin-mediated DNA loops reduce cis-element search space (Hsieh et al. 2022), or function to cyclically accumulate factors for promoter activation (Xiao et al. 2021), the relatively weak cohesin of C. elegans fits its underlying compact genome, in which enhancers are intragenic or immediately upstream of promoter (Reinke et al. 2013). The presence of fountains in C. elegans argues that preferential loading of cohesin at active enhancers is an evolutionarily conserved mechanism of 3D genome regulation regardless of genome size and CTCF presence. Future studies should address how cohesin-mediated organization of enhancer contacts contributes to cell type–specific gene regulation in the context of a compact animal genome.
Methods
Worm strain and growth
Worms were grown and maintained at 20°C–22°C on nematode growth medium (NGM) plates containing Escherichia coli strains OP50-1 and/or HB101. To isolate synchronized L2/L3 worms, gravid adults were bleached in 0.5 M NaOH and 1.2% bleach, and embryos were hatched overnight in M9 buffer (22 mM KH2PO4 monobasic, 42.3 mM Na2HPO4, 85.6 mM NaCl, 1 mM MgSO4). The resulting starved L1s were grown for 24 h at 22°C. Degron-GFP-tagged alleles were produced by SUNY Biotech and crossed to a CA1200 strain expressing TIR1 under the control of eft-3 promoter to produce ERC102 syb5520 [smc-3::GGGGS::AID::emGFP] III, [eft-3p::TIR1::mRuby::unc-54 3′UTR + Cbr-unc-119(+)] II, ERC103 syb6035 [WAPL-1::GGGGS::AID::emGFP] IV, [eft-3p::TIR1::mRuby::unc-54 3′UTR + Cbr-unc-119(+)] II, and ERC107 syb8826[scc-2::GGGGS::AID], ieSi57 II; unc-119(ed3) III. L2/L3 worms were used to minimize the potential effect of mitosis in somatic genome organization in our experiments. Although some germline proliferation is present, we isolate the function of the protein in the interphase by acutely depleting the protein of interest only in the somatic cells. Furthermore, somatic cells of L2/L3 worms divided and spent more time in the interphase compared with those of L1 worms. Thus, depleting cohesin in later larval development can reduce the “after-effect,” as chromosomes even after mitosis are in a constant state of flux (Nagano et al. 2017).
Auxin treatment
Auxin (indole-3-acetic-acid, Fisher 87-51-4) was resuspended in 100% ethanol to a concentration of 400 mM. Plates were prepared by adding resuspended auxin at a concentration of 1 mM to NGM media before pouring. Synchronized L2/L3 worms were washed three times with M9 and split into two. Half of the worms were transferred to NGM 10 cm plates containing 1 mM of auxin. The other half were placed in normal NGM 10 cm plates (no-auxin control). A maximum of 300 μL of settled worms was placed in one 10 cm plate. Worms were then washed one time with M9 and processed according to the future application. For ChIP and Hi-C, worms were cross-linked in 2% formaldehyde for 30 min, followed by quenching in 125 mM glycine for 5 min, and washing once with M9, and washing twice with PBS, PMSF, and protease inhibitors. For RNA-seq, worms were stored in TRIzol.
Microscopy
The worms were washed off the plate in M9 buffer, cross-linked in 0.5% formaldehyde for 15 min, and “freeze-cracked” by submerging in liquid nitrogen for 1 min and then thawing in a 37°C water bath. The worms were washed twice using 70% ethanol followed by a third wash using the wash buffer (PBS-0.05% Tween-20). The worms were resuspended in 1 mL of wash buffer and 1 μL of 2 μg/mL DAPI (FisherEN62248) and were incubated for 30 min in a 37°C water bath. The worms were then washed twice in PBS and imaged on slides for DAPI and GFP.
ChIP-seq
Two biological replicates with matching input samples were performed for each experiment. Around 100 μL of L2/L3 larvae pellet were dounce-homogenized with 30 strokes in FA buffer (50 mM HEPES/KOH at pH 7.5, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 150 mM NaCl) supplemented with PMSF and protease inhibitors (Calbiochem 539131). Dounced worms were sonicated in 0.1% sarkosyl for 15 min using a Picoruptor to obtain chromatin fragments between 200 and 800 bp. The protein concentration was determined using a Bradford assay (Bio-Rad 500-0006). One to two milligrams of protein extract was used per ChIP, and 5% was taken out to use as input DNA. The remaining protein extract was incubated with 3 to 10 μg of antibody rotating overnight at 4°C in a volume of 440 μL. Forty microliters of (1:1 V) Protein A and/or G-Sepharose beads that were previously washed three times with FA buffer were added and incubated, rotating for 2 h at 4°C. Beads were washed with 1 mL of each of the following buffers: two times with FA buffer, one time with FA-1 mM NaCl buffer, one time with FA-500 mM NaCl buffer, one time with TEL buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl at pH 8.0), and two times with TE buffer. Immunoprecipitated chromatin was eluted from beads by incubating in ChIP elution buffer (1% SDS, 250 mM NaCl, 10 mM Tris at pH 8.0, 1 mM EDTA) for 30 min at 65°C, treated with Proteinase K, and reverse cross-linked overnight at 65°C. Half of the ChIP DNA and 30 ng of input DNA were used for library preparation. End repair was performed in T4 ligase reaction buffer (New England Biolabs [NEB]), 0.4 mM of dNTPs, 20 U of T4 polynucleotide kinase (NEB), 3.5 U of large (Klenow) fragment (NEB), and 6 U of T4 DNA polymerase for 30 min at 20°C in a total volume of 33 μL. Reaction was cleaned using the Qiagen MinElute PCR purification kit. A-tailing reaction was performed in NEB buffer 2, 0.2 mM of dATPs, and 7.5 U of Klenow fragment-exo (NEB) for 60 min at 37°C in 25 μL. Reaction was cleaned using Qiagen MinElute PCR purification kit. Illumina TruSeq adapters were ligated to DNA fragments in a reaction containing 2× Quick ligation buffer (NEB), 0.25 μM of adapter, and 2 μL of Quick ligase (NEB) for 20 min at 23°C in 40 μL. The reaction was cleaned using Agencourt AMPure XP beads, and the eluted DNA was PCR-amplified in 50 μL using Phusion polymerase and TruSeq primers: forward, AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACG∗A; reverse, CAAGCAGAAGACGGCATACGAGA∗T, where ∗ represents a phosphorothioate bond. PCR reactions were cleaned using the Qiagen MinElute PCR purification kit. The eluted DNA was run on a 1.5% agarose gel, and fragments between 250 and 600 bp were gel-extracted using the Qiagen gel extraction kit. The library concentration was determined using the KAPA Library quantification kit. Single-end 75 bp sequencing was performed using the Illumina NextSeq 500.
For ChIP-seq data analysis, Bowtie 2 version 2.4.2 was used to align 75 bp single-end reads to WS220 with default parameters (Langmead and Salzberg 2012). BAM sorting and indexing were performed using SAMtools version 1.11 (Danecek et al. 2021). The BamCompare tool in deepTools version 3.5.0 was used to normalize for the sequencing depth using CPM and to create ChIP-Input coverage with a bin size of 10 bp and 200 bp read extension (Ramirez-Gonzalez et al. 2012). Only reads with a minimum mapping quality of 20 were used, and mitochondrial DNA, PCR duplicates, and blacklisted regions were removed (Amemiya et al. 2019). The average coverage data were generated by averaging ChIP-input enrichment scores per 10 bp bins across the genome. ChIP-seq peaks were identified using MACS2 version 2.1.1 (https://github.com/macs3-project/MACS). First, the MACS2 predictd function was used to predict fragment size. Then peaks were called using MACS2 callpeak function with the following parameters: -g ce -q 0.05 –nomodel –extsize $predicted_fragment_size. Peaks were called on both from the individual replicates and from the merged BAM files of the replicates. The peaks shown below average ChIP-seq tracks are peaks called using the merged BAM files, whereas the peaks shown below individual replicates are peaks called using the corresponding replicate. Unless specified, peak-based analyses in the main figures use peaks called using the merged BAM files. We reproduced these results using a more stringent approach: For each peak called in the merged BAM file, we counted the number of times it overlapped with a peak that was called in replicates and only kept those that were present in more than half of the replicates (Supplemental Fig. S5E). Additionally, we reproduced the peak-based analyses in the main figures using individual replicates (Supplemental Fig. S5F). Thus, our conclusions based on the relative numbers and genomic distribution of peaks are robust to the stringency of peak calling strategy.
Hi-C
Two biological replicates were performed for each experiment. Cross-linked L2/L3 worms collected as described above were resuspended in 20 μL of PBS per 50 μL of worm pellet and then dripped into liquid nitrogen containing mortar. The worms were grounded with pestle until fine powder. Grounded worms were cross-linked again in 2% formaldehyde using the TC buffer as described by the Arima high-coverage Hi-C kit, which uses four four-base cutters, DpnII, HinfI, DdeI, and MseI. The Arima manufacturer's protocol was followed, including the recommended method of the library preparation using KAPA hyperprep kit. Paired-end 100 bp sequencing was performed using the Illumina NovaSeq 6000.
For Hi-C data analysis: The Hi-C data were mapped to the ce10 (WS220) reference genome using default parameters of the Juicer pipeline version 1.5.7 (Durand et al. 2016). The biological replicates were combined using juicer's mega.sh script. The mapping statistics from the inter_30.txt output file are provided. The inter_30.hic outputs were converted to COOL format using the hicConvertFormat of HiCExplorer version 3.6 (Ramírez et al. 2018; Wolff et al. 2018) in two steps using the following parameters: (1) ‐‐inputFormat hic, ‐‐outputFormat cool, (2) ‐‐inputFormat cool ‐‐outputFormat cool ‐‐load_raw_values. The COOL file was balanced using cooler version 0.8.11 using the parameters: ‐‐max-iters 500, ‐‐mad-max 5, ‐‐ignore-diags 2 (Abdennur and Mirny 2020). The balanced COOL file was used for all downstream analysis.
For computing log-binned P(s) and its log-derivative, cooltools version 0.4.0 (https://github.com/open2c/cooltools) was used. For visualizing ChIP-seq data with Hi-C data in Python, pyBigwig version 0.3.18 (https://github.com/deeptools/pyBigWig) was used.
For fountain identification, fountains must be (1) observed in control, (2) observed in WAPL-1-depletion conditions, and (3) show significant decrease upon cohesin depletion. We apply the tool implemented in cooltools (version 0.4.0), which was originally developed to identify elements whose interaction across is insulated, to instead identify elements whose interaction across is facilitated. Instead of using local minima, the local maxima were used. Peak prominence was calculated for local maxima to identify their strength relative to the surrounding. Then otsu's threshold was used to remove local maxima that arise from random noise. We identified 449 (criteria 1) and 587 (criteria 2) local maxima in the control condition and the WAPL-1-depletion condition. To identify elements whose interaction across is significantly lost upon cohesin depletion, we used insulation score delta (control minus SMC3 depletion) to identify local maxima. The identified 831 (criteria 3) significant local maxima were further subsetted based on their overlap with criteria 1 and 2, resulting in the final 287 fountains (Supplemental Fig. S1C,D). The “fountain strength” is the peak prominence value of insulation delta (control minus SMC3 depletion). We reason that this metric best captures how strongly the interaction across the element is facilitated by cohesin.
Fontanka (Galitsyna et al. 2023; https://github.com/agalitsyna/fontanka) was used with the following steps. First, for snippet extraction, we manually identified three different types of shapes based on the tilt of the fountain: center, left, and right. We manually selected 10 snippets that appeared to show left or right tilt. For each shape, the 10 snippets were used to generate observed-over-expected on-diagonal pile-up matrices with flanking ±100 kb window. This pile-up is used as the “reference mask” to which the snippets from the rest of the genome is compared. Second, for the fountain score and Scharr score, we used the fontanka slice-windows function to generate genome-wide snippets using the parameter -W 100_000 for both the control condition and WAPL-1-depletion conditions using a 2 kb binned matrix. The fountain and Scharr scores for each window were calculated by fontanka-apply-fountain-mask. Third, for peak calling and thresholding, we applied otsu thresholding method on peak prominence returned from the fontanka-apply-fountain-mask function and applied a minimum threshold of 0.2 for fountain score. The Scharr score, which measures the uniformity/sharpness of the Hi-C matrix, can be used to infer rearrangements and incomplete reference genome data. Windows of Hi-C matrix with sharp transitions, which often arises from imperfect reference genome (rearrangements and missing segments), have a higher Scharr score, whereas windows containing smooth transitions in contact scores have a lower Scharr score. We only removed the top-scoring 5% of candidate fountains (false positives owing to reference genome) based on previous work showing that long-read assembly of C. elegans genome only added 1.8 Mb (<2%) to the existing reference genome (Yoshimura et al. 2019). Fourth, to leverage the WAPL-1 condition, we kept control fountains that overlapped (±10 kb) with those called in the WAPL-1 depletion, which shows sharper contrast in fountain shapes. Fifth, for validation, we ensured that the average on-diagonal pile-up of the called fountains shows distinct shapes.
mRNA-seq
L3 larvae were collected and stored in TRIzol (Invitrogen) at −70°C. Total RNA was extracted through the freeze-crack method and processed as described previously (Albritton et al. 2014). RNA was cleaned up using the Qiagen RNeasy MinElute cleanup kit. mRNAs were purified using Sera-Mag Oligo (dT) beads (Thermo Scientific), fragmented using Ambion (AM8740), and cleaned using the Qiagen RNAeasy minElute kit. RNA was reverse-transcribed using the Superscript III system (Invitrogen 18080-051). Residual dNTP was removed using ethanol precipitation. The second strand was synthesized using DNA polymerase I (Invitrogen). dsDNA was purified using the Qiagen DNA minElute kit. Single-stranded illumine libraries were prepared as indicated for ChIP-seq with the following modification: Prior to PCR amplification, uridine was digested using uracil-N-glycosylase (Thermo Scientific EN0361).
For mRNA-seq data analysis, single-end 75 bp sequencing was performed using the Illumina NextSeq 500. Reads were mapped to the WS220 (ce10) C. elegans genome using HISAT2 version 2.2.1 (Kim et al. 2015) with the parameter ‐‐rna-strandness R. The SAM output from HISAT2 and WS220 GTF files was used to calculate counts per gene using HTSeq version 0.13.5 (Anders et al. 2015). The raw counts were normalized FPKM using cufflinks version 2.2.1 (Roberts et al. 2011), and then FPKM was converted to TPM. For comparing between conditions, the raw counts were used for the R package DESeq2 version 1.30.0 (Love et al. 2014).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE237663. A list of data and statistics are provided in Supplemental File 1. Analysis scripts are available at GitHub (https://github.com/ercanlab/2023_Kim_cohesin) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Research in this manuscript was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number R35 GM130311. J.K. was partially supported by NIGMS Predoctoral Fellowship T32HD007520. We thank Dominic Balcon for testing cohesin antibodies and GenCore at the NYU Center for Genomics and Systems Biology for sequencing and raw data processing.
Author contributions: J.K. and S.E. conceptualized the project and designed experiments. J.K. and H.W. performed experiments. J.K. performed data analysis. J.K. and S.E. prepared figures and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279365.124.
- Received March 18, 2024.
- Accepted February 15, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








