
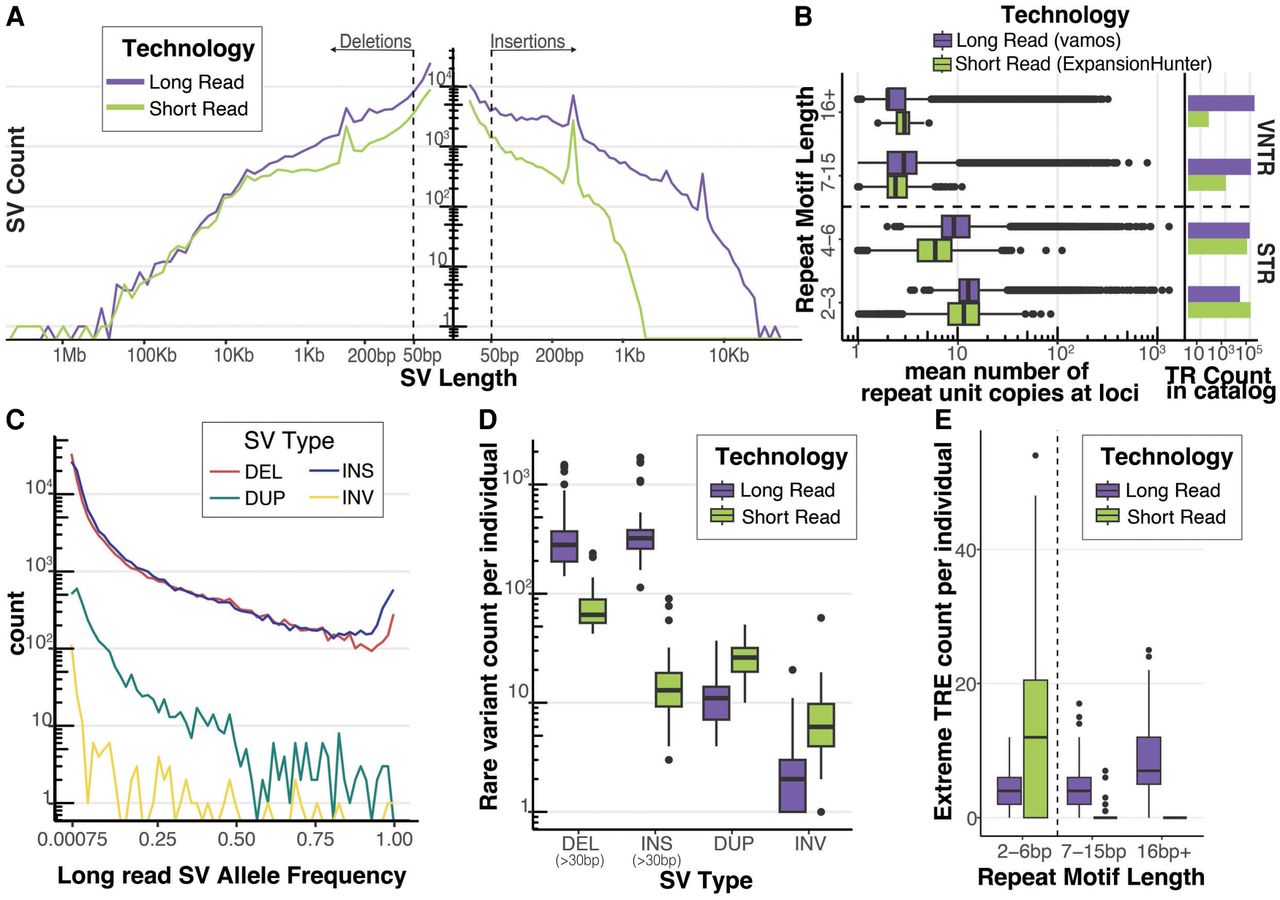
Long-read sequencing detects rare SVs and extreme tandem repeat expansions (TREs). (A) Length distribution of deletions and insertions detected by each technology on a log–log axis. SVs were called with a consensus SV calling pipeline including SVIM, cuteSV, and Sniffles2 for long reads and MantaSV calls were genotyped with paragraph for short reads. Dashed line represents 50 bp, the threshold for calling an indel an SV. (B) Mean tandem repeat copy numbers estimated from the UDN genomes stratified by repeat motif length. Short tandem repeats (STR) have repeat motifs between 2 bp and 6 bp. Variable number tandem repeats (VNTRs) have repeat motifs greater or equal to 7 bp. Vamos was used to genotype tandem repeat copy number in long reads and ExpansionHunter was used in short reads. Each tool used a different tandem repeat loci catalog to define TRs. Counts of TRs by repeat motif length bins present in the tools respective catalog is also plotted. (C) Allele frequency distribution of long-read discovered SVs from Jasmine-SV merge with ADRC genomes. ADRC provided a reference sample of 600 nanopore genomes to allow robust estimation of minor allele frequencies. (D) Count of rare SVs (MAF < 0.01), detected per individual stratified by SV Type and Technology. Short-read SVs were annotated with allele frequencies using SVAFotate and a lookup in gnomAD, CCDG, and 1000 G. (E) Count of extreme TRE detected per individual. Extreme TRE outliers in each technology were called by jointly estimating repeat copy number distribution of long-read vamos calls with the ADRC and of short-read ExpansionHunter calls with 1000 G, and then calculating for each allele its average distance from its k-nearest neighbors. Extreme TREs were defined as alleles with a standardized mean neighbor distance (MND) >2, with k = 5 for long reads and k = 25 for short reads.








