
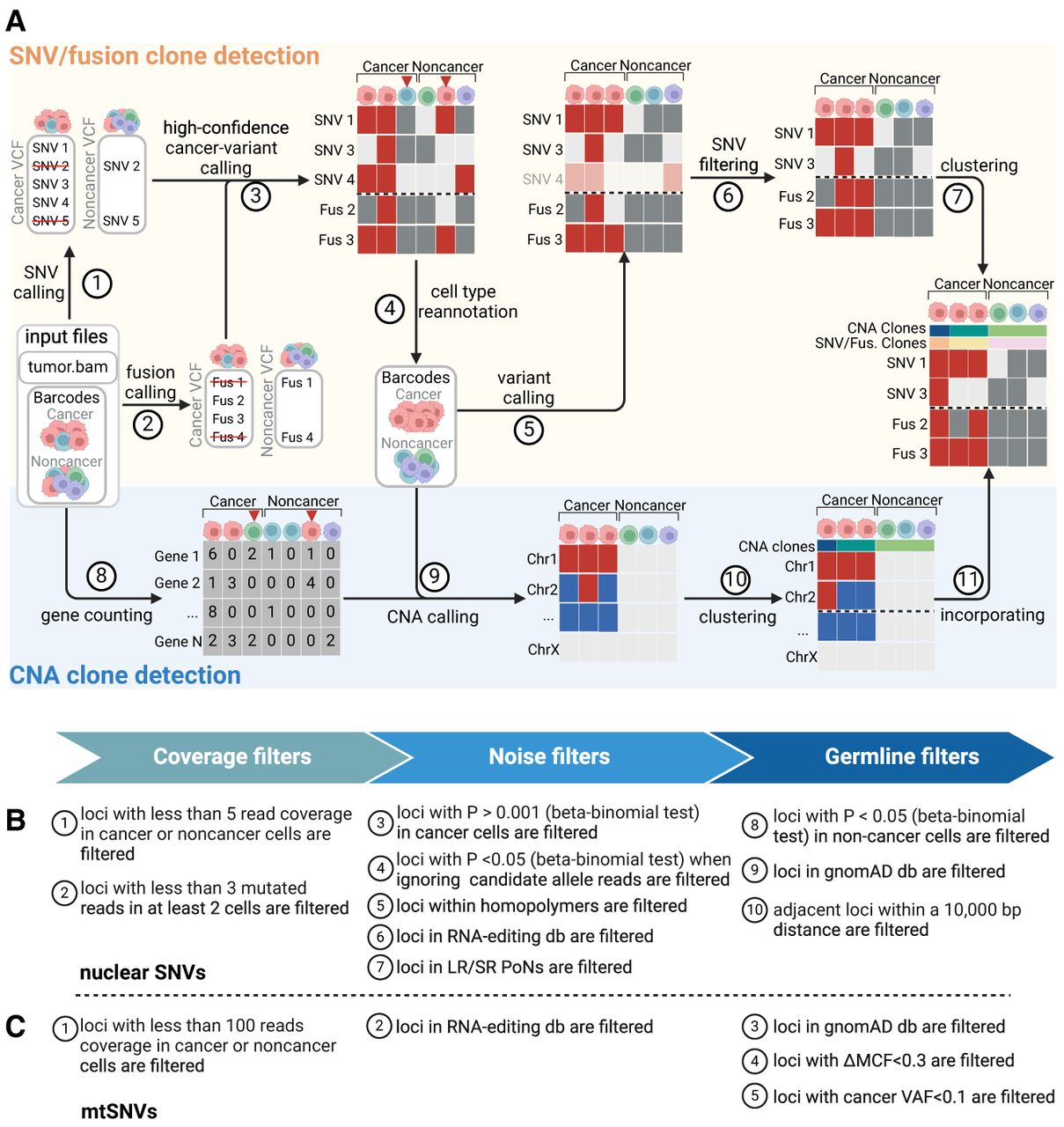
Overview of LongSom. (A) LongSom's methodology for detecting somatic SNVs, fusions, and CNAs and subsequently inferring cancer subclones in LR scRNA-seq individual patients data. (1) SNV and (2) fusion candidates are detected from pseudo-bulk samples. (3) High-confidence cancer variants (SNVs and fusions) are selected based on mutated cell fraction in cancer and noncancer cells. (4) Cells are reannotated based on high-confidence cancer variants. (5) A new set of candidate variants is called based on reannotated barcodes. (6) Candidate SNVs are filtered through a set of 10 filters. (7) Cells are clustered based on somatic fusions and SNVs. In parallel, (8) gene expression per cell is computed, (9) CNAs are detected, (10) cells are clustered based on CNAs, and (11) CNA clones are incorporated into the fusions and SNVs clustered matrix. (B) Candidate nuclear SNV successive filtering steps. Candidates passing all 10 steps are called as somatic SNVs (Methods). (C) Candidate mtSNVs filtering steps. ΔMCF represents the difference of mutated cell fractions (MCFs) between cancer and noncancer cells. Candidates passing all five steps are called as somatic mtSNVs (Methods).








