
Abstract
As the number and variety of assembled genomes continue to grow, the number of annotated genomes is falling behind, particularly for eukaryotes. DNA-based mapping tools help to address this challenge, but they are only able to transfer annotation between closely related species. Here we introduce LiftOn, a homology-based software tool that integrates DNA and protein alignments to enhance the accuracy of genome-scale annotation and to allow mapping between relatively distant species. LiftOn's protein-centric algorithm considers both types of alignments, chooses optimal open reading frames, resolves overlapping gene loci, and finds additional gene copies when they exist. LiftOn can reliably transfer annotation between genomes representing members of the same species, as we demonstrate on human, mouse, honeybee, rice, and Arabidopsis thaliana. It can further map annotation effectively across species pairs as far apart as mouse and rat or Drosophila melanogaster and Drosophila erecta.
Recent advancements in sequencing technologies have resulted in an exponential increase in available genome assemblies. Long-read sequencing technology, which has become faster, more cost-effective, and more accurate over the past decade (Shendure et al. 2017; Van Dijk et al. 2018; Logsdon et al. 2020; Kovaka et al. 2023; Marx 2023), has notably enhanced the accuracy and contiguity of genome assemblies. A prominent example is the telomere-to-telomere (T2T) assembly of the human genome CHM13 (Nurk et al. 2022), which has subsequently been used to guide the assembly of multiple additional human genomes (Shumate et al. 2020; Zimin et al. 2022; Chao et al. 2023). As of September 2024, the NCBI Genome Assembly database (https://www.ncbi.nlm.nih.gov/datasets/genome/) contains 2.48 million genome assemblies, of which 43,120 are eukaryotes, 2.20 million are bacteria, 24,050 are archaea, and 192,600 are viruses.
Genome annotation—identifying genes and other biological features—is essential to understanding the biology of these genomes (Stein 2001). Annotating eukaryotic genomes presents particular challenges (Danchin et al. 2018; Salzberg 2019) owing to their multiexon gene structures, extensive intergenic regions, and sparse gene density. This complexity necessitates detailed analysis and manual curation, making the process slower and more difficult to automate than genome assembly (Yandell and Ence 2012; O'Leary et al. 2016; Frankish et al. 2019; Salzberg 2019). The most widely used approaches to annotation are software pipelines such as MAKER, MAKER-P (Cantarel et al. 2008; Campbell et al. 2014), BRAKER, and BRAKER2 (Hoff et al. 2019; Brůna et al. 2021), which combine ab initio prediction, protein alignments, and other evidence to create annotation at the genome scale, although these systems primarily annotate protein-coding genes. RNA sequencing (RNA-seq) can more accurately identify exon–intron structure because it directly captures gene transcripts after splicing has removed introns, although RNA-seq may miss genes that are expressed at low levels or in hard-to-capture tissues (Lonsdale et al. 2013; Weinstein et al. 2013). In contrast, annotation mapping or “lift-over” strategies, which transfer annotations from a well-annotated genome to a newly sequenced one based on homology and synteny, provide a more efficient and cost-effective annotation method, particularly when annotations from the same or closely related species are available (Sharma et al. 2017; Fiddes et al. 2018; Shumate and Salzberg 2021).
Currently, the best approach for transferring annotation between assemblies is DNA-based, exemplified by the Liftoff (Shumate and Salzberg 2021) and CAT (Fiddes et al. 2018) programs, which were used to create the initial annotation of the human T2T-CHM13 genome (Nurk et al. 2022) based on the annotation of the human reference genome, GRCh38. However, in cases in which the DNA sequence of a newly assembled genome deviates substantially from the reference genome, a DNA-based alignment process sometimes produces transcripts with invalid open reading frames (ORFs) or with erroneous splice sites. DNA-based mapping becomes even more challenging when the new genome is less closely related to the available reference.
By comparison with DNA, protein sequences of orthologous genes are conserved at much greater evolutionary distances. This observation motivated us to develop a new method that integrates protein sequence alignment into the lift-over process. A key step in this approach is to align protein sequences from the reference genome to the target, considering all six reading frames and allowing for spliced alignments to span introns. However, protein-based alignment alone cannot map all annotation between two genomes. First and most obviously, it cannot capture untranslated regions (UTRs) on either end of a transcript. Second, when small exons are separated by much longer introns, as is common in the human genome, protein-based aligners may miss some small exons entirely (as illustrated in Supplemental Fig. S1A,B). Third, because intronic alignments are not considered, protein-to-DNA alignment is susceptible to aligning proteins to pseudogenes. Fourth, this approach sometimes combines coding sequences (CDSs) from distinct genes when multiple members of a gene family are arranged in tandem along a genome (Supplemental Fig. S1C). Finally, another obvious limitation is that a protein alignment strategy cannot transfer annotations of noncoding genes or other features.
LiftOn is a homology-based genome annotation tool that uses both DNA and protein sequence alignment and that builds on Liftoff, the current leading homology-based annotation lift-over tool. LiftOn includes several key functions to create annotation: (1) It uses a protein-maximization (PM) algorithm that combines both DNA and protein sequence alignment to generate protein-coding gene annotations that maximize similarity to the reference proteins; (2) it checks alternative ORFs for truncated proteins to identify the ORF that yields the longest match to the reference protein; (3) similarly to Liftoff, it finds extra copies of protein-coding and noncoding gene copies in the target genome; (4) it reports the various types of mutations for proteins that fail to match the reference perfectly, similarly to LiftoffTools (Shumate and Salzberg 2022); and (5) it resolves issues such as overlapping gene loci and multimapping for genes within a large gene family. By combining the advantages of DNA and protein sequence alignment, LiftOn generates better protein-coding gene annotation than either alignment method can achieve on its own.
Results
In our experiments below, we demonstrate how LiftOn can yield substantial improvements in mapping annotations from one human genome, GRCh38, to another, T2T-CHM13, using three different human annotation sets: MANE (note that MANE is a high-quality annotation with one transcript for each protein-coding gene, and all MANE transcripts are contained within RefSeq and CHESS) (Morales et al. 2022), CHESS (Varabyou et al. 2023), and RefSeq (Pruitt et al. 2007). Additionally, we show that LiftOn is effective for mapping annotation between members of nonhuman species, including Mus musculus (mouse), Apis mellifera (honey bee), Arabidopsis thaliana (thale cress), and Oryza sativa (rice). To demonstrate LiftOn's effectiveness at mapping annotation between distinct but closely related species, we mapped human genes onto Pan troglodytes (chimpanzee). Finally, we illustrate that LiftOn works on more distantly related species by mapping annotation from Drosophila melanogaster to Drosophila erecta and from M. musculus to Rattus norvegicus.
A two-step PM algorithm to improve protein-coding gene annotations
LiftOn implements a two-step PM algorithm (illustrated in Fig. 1 and discussed in more detail in Methods) to find the best annotations at protein-coding gene loci. First, it uses a chaining algorithm, described below, to find the exon–intron boundaries of protein-coding transcripts. Second, if the CDS does not preserve the entire protein sequence of the reference protein, it adjusts the CDS boundaries to preserve as much of the reference protein as possible (note that CDS features are defined as the coding portions of exons, from start to stop, and excluding untranslated portions of exons).
The protein-maximization (PM) algorithm consists of two modules: (A–E) the chaining algorithm and (F–K) the open reading frame (ORF) search algorithm. (A) Matched protein-coding transcripts mapped by Liftoff (green) and miniprot (orange) at the same location in a target genome. The transcript in blue represents the correct transcript annotation on the target genome. Liftoff's mapping has an erroneous splice junction between L3 and L4, whereas miniprot's mapping has a missing splice junction in M6. (B) Pairwise alignment results of the proteins mapped by Liftoff and miniprot to the reference protein. The figure shows a premature stop codon in the Liftoff protein, and the miniprot alignment has a mismatched protein sequence at the end. (C) Pairwise alignment mappings with added exon/CDS boundaries. (D) CDSs are grouped based on the cumulative lengths of the amino acids in the reference protein as described in the main text. In this example, CDSs are organized into groups: , , , , , , , , , and . The chaining algorithm iterates through each group, comparing the corresponding partial protein sequences to the reference protein and chaining those with higher protein sequence identity. (E) In this example, , , , , and are chained, forming the new protein-coding transcript CDS list. This list includes L1, L2, M3, M4, L5, L6, and L7 in the LiftOn annotation. (F–K) Schematic diagrams illustrating how the ORF search algorithm handles various types of sequence mutations. This process leads to changes in the gene annotation of both translated and untranslated regions (UTRs). (F) A frameshift mutation is a variation caused by the insertion or deletion of a sequence of nucleotides whose length is not divisible by three. In this example, the indel introduces a premature stop codon. (G,H) Point mutations leading to premature stop codons. LiftOn searches for the longest ORF, considering two scenarios: G depicts the selection of the first encountered stop codon, and H illustrates the switch to the downstream start codon. (I) Stop codon loss. When a stop codon is deleted, LiftOn identifies a new stop codon in the 3′ UTR. (J,K) Start codon loss. In this scenario, LiftOn searches for a new start codon, exploring both downstream in the coding region (J) and upstream in the 5′ UTR (K).
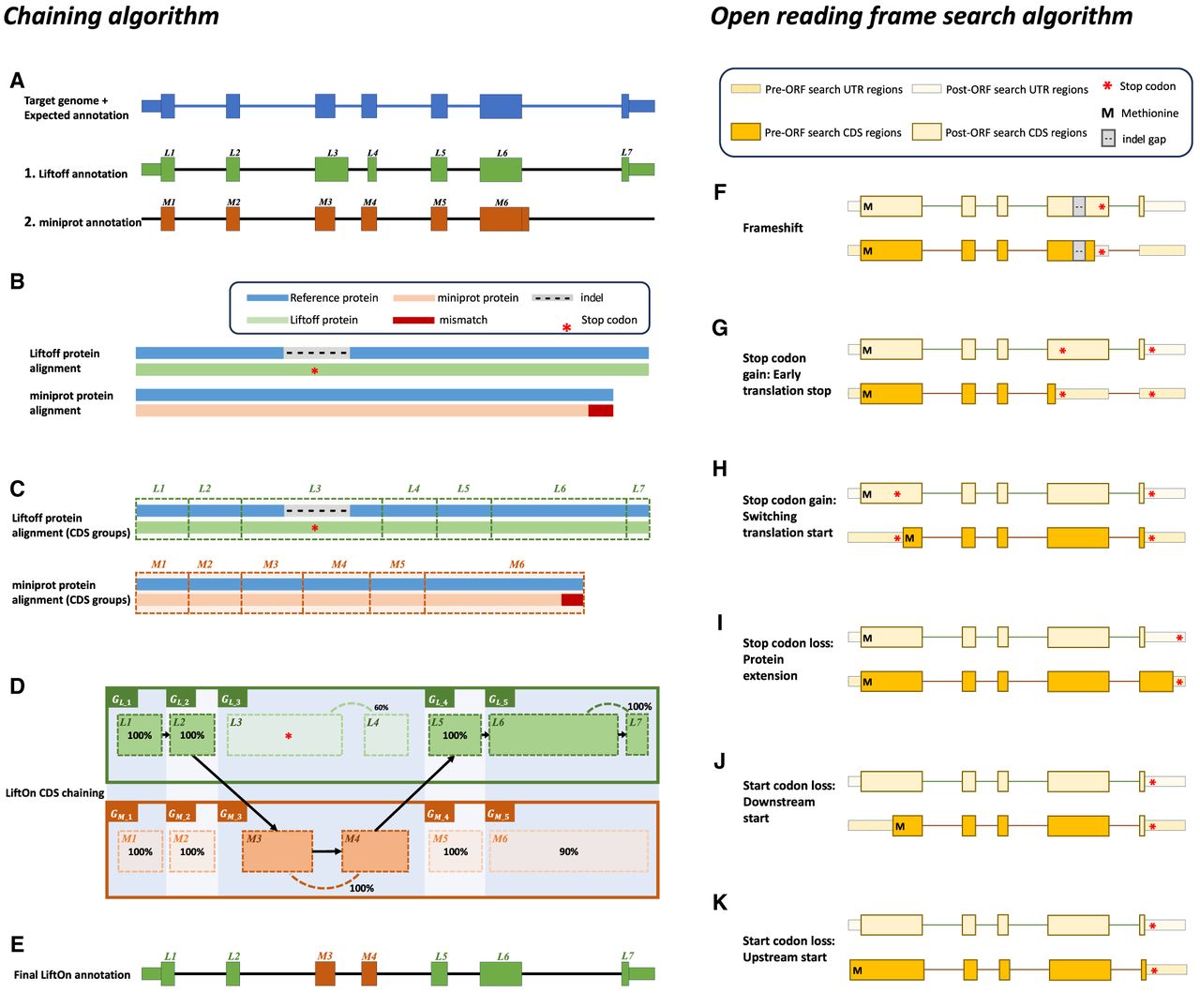
The chaining algorithm (Fig. 1A–E) starts by pairing up miniprot (Li 2023) alignments with transcripts identified by Liftoff (for details on the pairing approach, see Methods and Algorithm S1). After two transcripts are paired, the protein sequences from the Liftoff and miniprot annotations are then aligned to the full-length reference protein, as illustrated in Figure 1B. Subsequently, LiftOn maps the CDS boundaries from both the Liftoff and miniprot annotations onto the protein alignment (Fig. 1C; Algorithm S2).
The CDSs within the Liftoff and miniprot annotations are grouped in the 5′ to 3′ direction. The CDS groups are represented as and , respectively, for LiftOff and miniprot, where i denotes the ith group in that annotation (Fig. 1D).
The grouping process begins with the first CDS in each annotation and continues until reaching the endpoints of the downstream CDS in Liftoff and miniprot, where the number of aligned amino acids from the reference protein is equal. This forms the first CDS group in Liftoff, denoted as , and the first CDS group in miniprot, denoted as . Subsequent groups start from the previous endpoint in both Liftoff and miniprot, extending until the number of aligned amino acids from the reference protein matches for both annotations again. These subsequent groups are represented as and , respectively. The grouping process concludes upon reaching the last CDS in both annotations (for more details, see Algorithm S3).
Within each group, or , we calculate the partial protein sequence identity and select the group with higher protein sequence identity score (Fig. 1D; Algorithm S4). In case of a tie, LiftOn prioritizes the Liftoff annotation, , so that it will include UTRs in its output. The selected group of CDSs is represented by . All CDSs in are then concatenated to form the final LiftOn transcript, as shown in Figure 1E. This transcript is an ordered sequence of CDSs sourced from either Liftoff or miniprot, with the goal of maximizing protein similarity to the reference protein. This approach is particularly effective in addressing issues such as in-frame indels or mis-splicing that may arise from misalignments, as illustrated in Figure 1A.
After applying the chaining algorithm, LiftOn attempts to make further improvements in the CDS regions, as illustrated in Figure 1, F–K. It searches the translations of protein-coding transcripts and adjusts CDS boundaries to avoid early stop codons (Fig. 1F,G), choose better translation start sites (Fig. 1H,J,K), or extend proteins with stop codon loss (Fig. 1I). Supplemental Figure S2 provides additional Integrative Genomics Viewer (IGV) (Robinson et al. 2011; Thorvaldsdóttir et al. 2013) screenshots that illustrate LiftOn's results for each scenario. After making these adjustments, LiftOn evaluates the differences between the reference and target transcripts and, similarly to LiftoffTools (Shumate and Salzberg 2022), produces a mutation report. Transcripts are deemed “identical” when their target and reference gene DNA sequences are entirely the same. For nonidentical sequences, LiftOn categorizes their differences using these categories: synonymous, nonsynonymous, in-frame insertion, in-frame deletion, frameshift, stop codon gain, stop codon loss, and start codon loss.
Lifton improves human annotation lift-over
For our first experiment, we used human annotation from RefSeq release 220 (GCF_000001405.40-RS_2023_03) (Pruitt et al. 2007) as the reference annotation to map annotations from the GRCh38 onto the T2T-CHM13 (Supplemental Fig. S3). In total, the reference annotation contained 37,986 protein-coding and noncoding genes and 160,561 transcripts, of which 130,528 were protein-coding (for all mapped genes, see Supplemental Table S1; for our gene counting approach, see Methods). We focused our analysis on the protein-coding transcripts because those are the ones for which LiftOn can produce improvements over DNA-based methods. At the gene level, LiftOn successfully lifted over 37,828 genes, whereas 158 genes remained unmapped, of which 103 were protein-coding (Supplemental Table S2). The overall gene mapping rate was 99.6%. All 158 genes mapped to the locations of other genes on the target genome, likely owing to copy number variation. Out of the successfully mapped genes, 37,453 (19,738 protein-coding and 17,715 noncoding) were mapped as single copies, and 375 genes (86 protein-coding and 289 noncoding) were mapped with extra copies. In total, 38,886 gene loci were mapped onto T2T-CHM13 (Table 1).
Statistics for LiftOn at both the gene and transcript levels, as a result of mapping RefSeq release 220 annotation from the GRCh38 human genome to T2T-CHM13
| Total feature count | Protein-coding feature count | Noncoding feature count | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Single copy | Extra copy | Extra copy count | Total | Single copy | Extra copy | Extra copy count | Total | ||
| Gene | |||||||||
| Reference (GRCh38) | 37,986 | 19,927 | 18,059 | ||||||
| Target (CHM13) | 38,916 | 19,738 | 86 | 320 | 20,144 | 17,715 | 289 | 768 | 18,772 |
| Transcript | |||||||||
| Reference (GRCh38) | 160,561 | 130,528 | 30,033 | ||||||
| Target (CHM13) | 161,701 | 129,967 | 239 | 573 | 130,779 | 29,488 | 410 | 1024 | 30,922 |
[i] Extra copy refers to the number of genes with extra copies; extra copy count refers to the total number of additional copies for those extra copy genes.
In assessing overall protein-coding gene annotations, we computed gap-compressed protein sequence identity scores for LiftOn, Liftoff, and miniprot. Figure 2A compares the mapping of protein-coding genes using Liftoff, which is based on DNA alignment, to miniprot, which uses protein-to-DNA alignment. Dots in the lower right part of the panel indicate transcripts for which Liftoff was superior to miniprot, as measured by protein sequence identity, and the upper left part of the plot shows transcripts for which miniprot was superior. This comparison shows that neither approach dominates the other. In the LiftOn versus Liftoff comparison (Fig. 2B), 866 transcripts exhibit higher protein sequence identity, with 113 of those achieving 100% identity. Similarly, the LiftOn versus miniprot comparison (Fig. 2C) shows that LiftOn finds better matches for 30,266 protein-coding transcripts, improving 22,746 to 100% identity. The protein sequence identity distributions are shown in Supplemental Figure S4.
Comparative analysis of various tools for mapping RefSeq protein annotations from GRCh38 to T2T-CHM13 v2.0. (A–C) Scatter plots of protein sequence identity. (A) Comparison between miniprot (y-axis) and Liftoff (x-axis), (B) comparison between LiftOn (y-axis) and Liftoff (x-axis), and (C) comparison between LiftOn (y-axis) and miniprot (x-axis). (D–G) Examples of improved annotation owing to LiftOn's two-step PM algorithm. (D) LiftOn uses Liftoff's annotation to correct a splice junction missed by miniprot in transcript NM_001083965.2 of the TDRKH gene. (E) For transcript NM_001384763.1 of the SLC22A31 gene, LiftOn uses miniprot's annotation to resolve an incorrect acceptor site from Liftoff's annotation. (F) For transcript XM_011517662.4 of the WASHC1 gene, LiftOn combines both annotations to rectify an omitted CDS by miniprot and a misidentified splice junction by Liftoff between the fifth and sixth exons. (G) LiftOn's ORF search algorithm selects an alternative downstream start codon for a frameshift mutation in transcript NM_001004692.2 of the OR2T12 gene, thereby conserving the majority of the protein sequence.
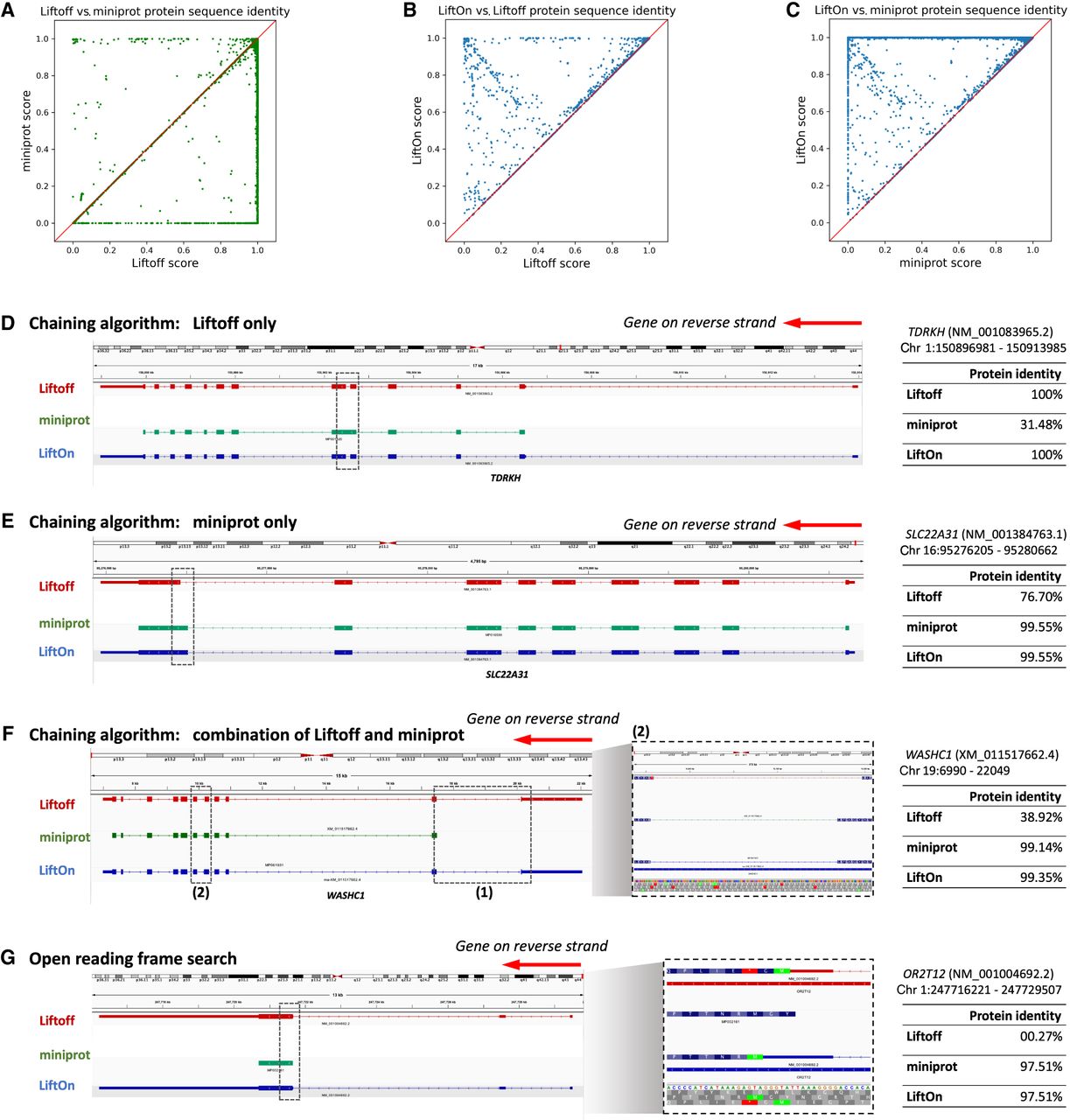
The LiftOn PM algorithm can improve protein-coding transcript annotations in the four scenarios illustrated in Figure 2. First, as observed with the TDRKH gene (Fig. 2D; Supplemental Fig. S5A), miniprot missed a splice junction, leading to the retention of an intron and a premature stop codon. Here LiftOn adopted Liftoff's CDS, which yielded the correct protein. Second, as shown in Figure 2E and Supplemental Figure S5B for the SLC22A31 gene, miniprot correctly identified an AG acceptor site that Liftoff missed, yielding the correct CDS, which LiftOn combined with the UTRs from Liftoff. Third, LiftOn sometimes was able to fix errors in both methods, as shown in Figure 2F and Supplemental Figure S5C for the gene WASHC1. In this example, LiftOn corrected the omission of the first coding exon by miniprot and fixed an incorrect splice junction found by Liftoff, one that erroneously introduced a premature stop codon between the fifth and sixth exons. LiftOn's chaining algorithm effectively consolidates these segments, improving the protein sequence identity to 99.4%. Fourth, as shown in Figure 2G for the OR2T12 gene, a mutation introduced a premature stop codon (Fig. 2G, inset), and the ORF search algorithm in LiftOn adjusted by scanning for ORFs that best match the reference protein sequence. Here, it found an alternative start codon just two codons downstream that maintained a near full-length match to the reference protein.
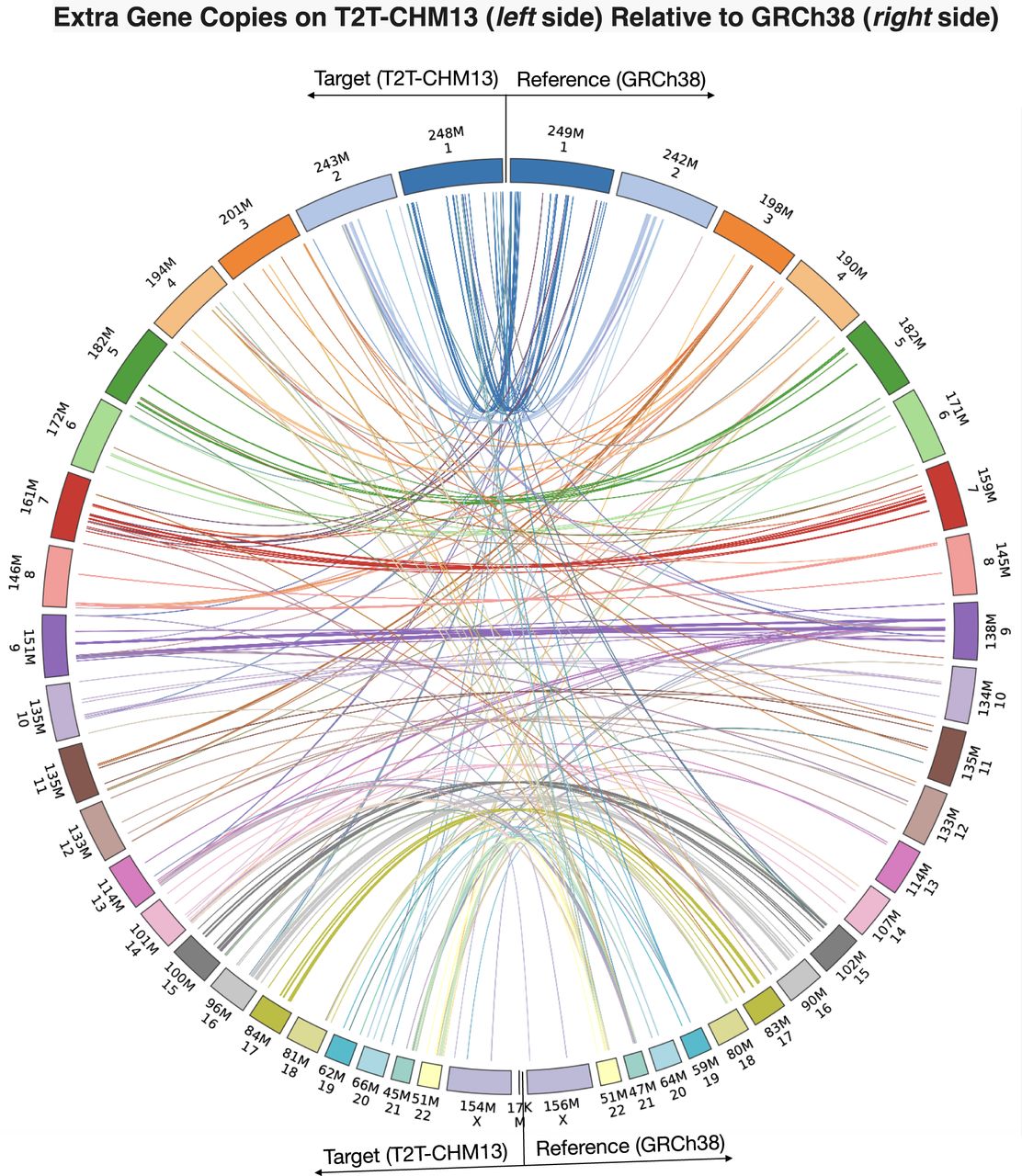
LiftOn identifies and reports additional copies of lifted-over features, similarly to Liftoff. In the mapping from GRCh38 to CHM13, LiftOn identified 86 protein-coding genes with at least one extra copy, for a total of 320 new protein-coding gene loci. Of these, 289 extra gene copies were detected by Liftoff and 31 by miniprot, detailed in Supplemental Tables S3 and S4. The Circos plot (Krzywinski et al. 2009) shows the relative positions of extra gene copies between the two genomes, illustrating that most were located on the same chromosomes (Fig. 3).
Plot illustrating the locations of extra gene copies found on T2T-CHM13 (left side) compared to GRCh38 (right side). The Circos plot was generated using pyCircos (https://github.com/ponnhide/pyCircos). Each line represents a gene copy mapped from the reference genome to the target genome, with colors indicating different chromosomes. The lines are color-coded by the chromosome of the original copy. The bands within the plot are sized proportionally to reflect the actual size of these chromosomes.
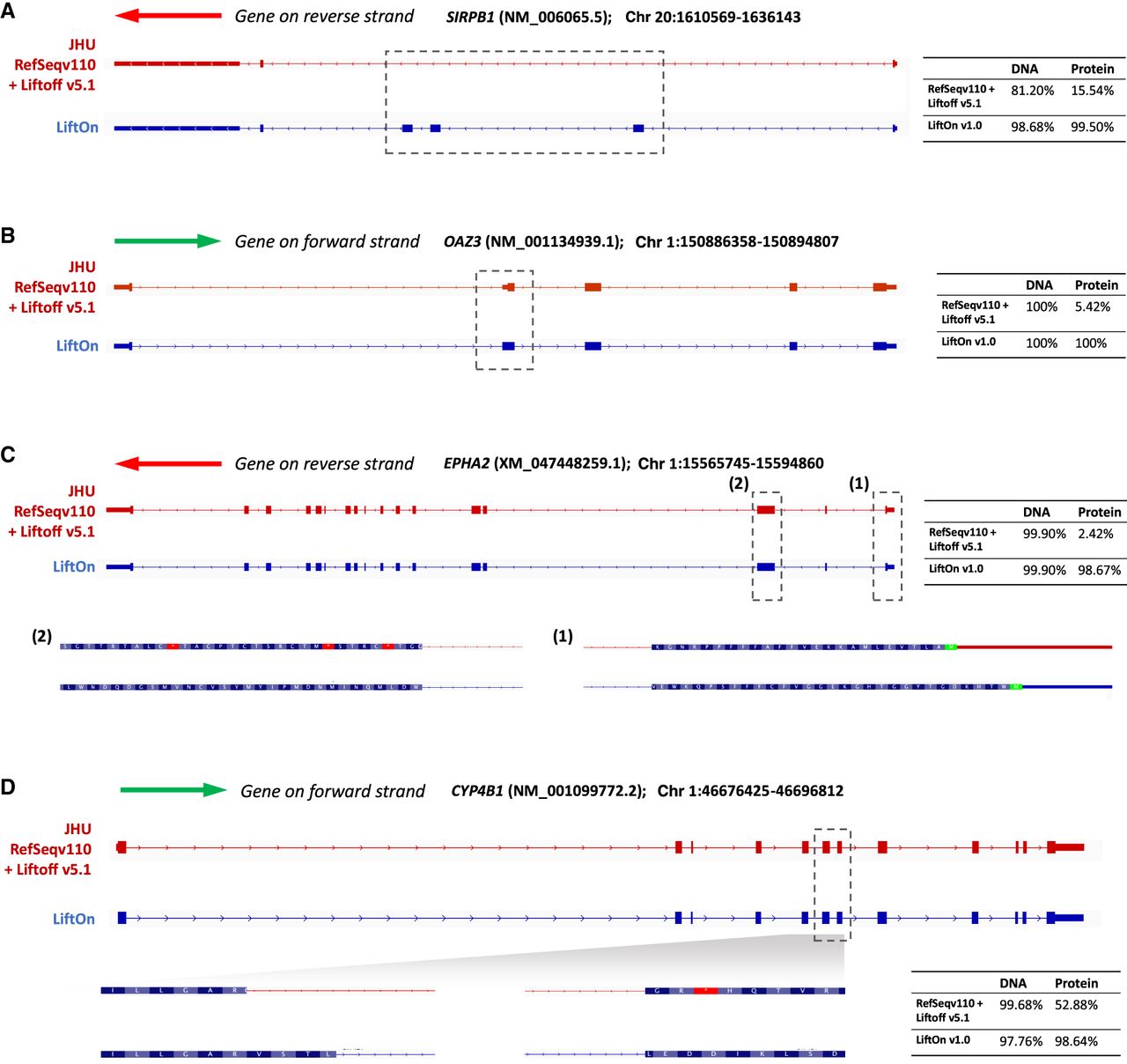
We further compared the LiftOn-generated annotation of CHM13 with the current release of the T2T-CHM13 annotation (available from the T2T GitHub site, https://github.com/marbl/CHM13; https://s3-us-west-2.amazonaws.com/human-pangenomics/T2T/CHM13/assemblies/annotation/chm13v2.0_RefSeq_Liftoff_v5.2.gff3.gz). We excluded the annotation of Chromosome Y from this analysis owing to the extremely complex repeat structure of that chromosome, which confounds most attempts to align genes consistently to it (Rhie et al. 2023). Our comparison considered only protein-coding transcripts on Chromosomes 1–22 and X. LiftOn's annotation contained 665 protein-coding transcripts that had a higher protein sequence identity to the corresponding proteins on GRCh38, compared with the current CHM13 annotation. Four examples are shown in Figure 4A–D, each of which illustrates how LiftOn can improve the fidelity of the match between the source protein and the mapped-over version.
Examples in which LiftOn improves the current T2T-CHM13 annotation. (A) In the NM_006065.5 transcript of the SIRPB1 gene, the current CHM13 annotation omits three coding exons. The LiftOn version finds those exons and increases the DNA sequence identity from 81% to 98%. (B) In transcript NM_001134939.1 of OAZ3, the CHM13 annotation incorporates a partial CDS in the second exon, leading to a truncated protein. LiftOn corrects this misannotation, increasing the protein sequence identity from 5.42% to 100%. (C) In transcript XM_047448259.1 of EPHA2, the published annotation chooses the wrong start codon. LiftOn finds a better start codon that improves the protein sequence identity from 2.4% to 98.7%. (D) In transcript NM_001099772.2 from CYP4B1, LiftOn shifts the donor site of the seventh coding exon by 11 nucleotides, fixing a frameshift and improving the protein sequence identity from 53% to 99%.
Among the 130,528 protein-coding transcripts mapped onto CHM13 by LiftOn, 13,486 transcripts were identical and 86,335 had only synonymous mutations. Of the remaining 30,707 transcripts, only 1046 had a protein with <95% identity to the reference protein. Overall, LiftOn's mapped-over annotation has greater fidelity to the source annotation (from GRCh38) than the current T2T-CHM13 annotation (Supplemental Fig. S6).
We observed similar improvements (compared with both Liftoff and miniprot) when we lifted over different human annotations, including MANE (Supplemental Note S1; Supplemental Table S5; Supplemental Fig. S7; Morales et al. 2022) and CHESS3 (Supplemental Note S2; Supplemental Table S6; Supplemental Fig. S8; Varabyou et al. 2023). Additionally, to assess the ability of LiftOn to map annotation between genomes of non-human species, we ran experiments on two different genomes from each of four plant and animal species—M. musculus (Supplemental Note S3; Supplemental Table S7; Supplemental Fig. S9; Chinwalla et al. 2002; Schmid-Siegert et al. 2023), A. mellifera (honeybee) (see Supplemental Note S4; Supplemental Table S8; Supplemental Fig. S10; Wallberg et al. 2019; Cao et al. 2021), O. sativa (Asian rice) (Supplemental Note S5; Supplemental Table S9; Supplemental Fig. S11; Kawahara et al. 2013; Shang et al. 2023), and A. thaliana (Supplemental Note S6; Supplemental Table S10; Supplemental Fig. S12; Lamesch et al. 2012; Naish et al. 2021)—and obtained comparable enhancements. These analyses demonstrate the robust performance of LiftOn in handling annotation lift-over across a broad spectrum of organisms.
LiftOn improves both closely and distantly related species annotation lift-over
To demonstrate that LiftOn can reliably lift-over cross-species annotations, we used it to transfer genes between both closely related and more distantly related species. We measured genomic distance using Dashing 2 (Baker and Langmead 2023) and Mash (Ondov et al. 2016).
Mapping from Homo sapiens to P. troglodytes
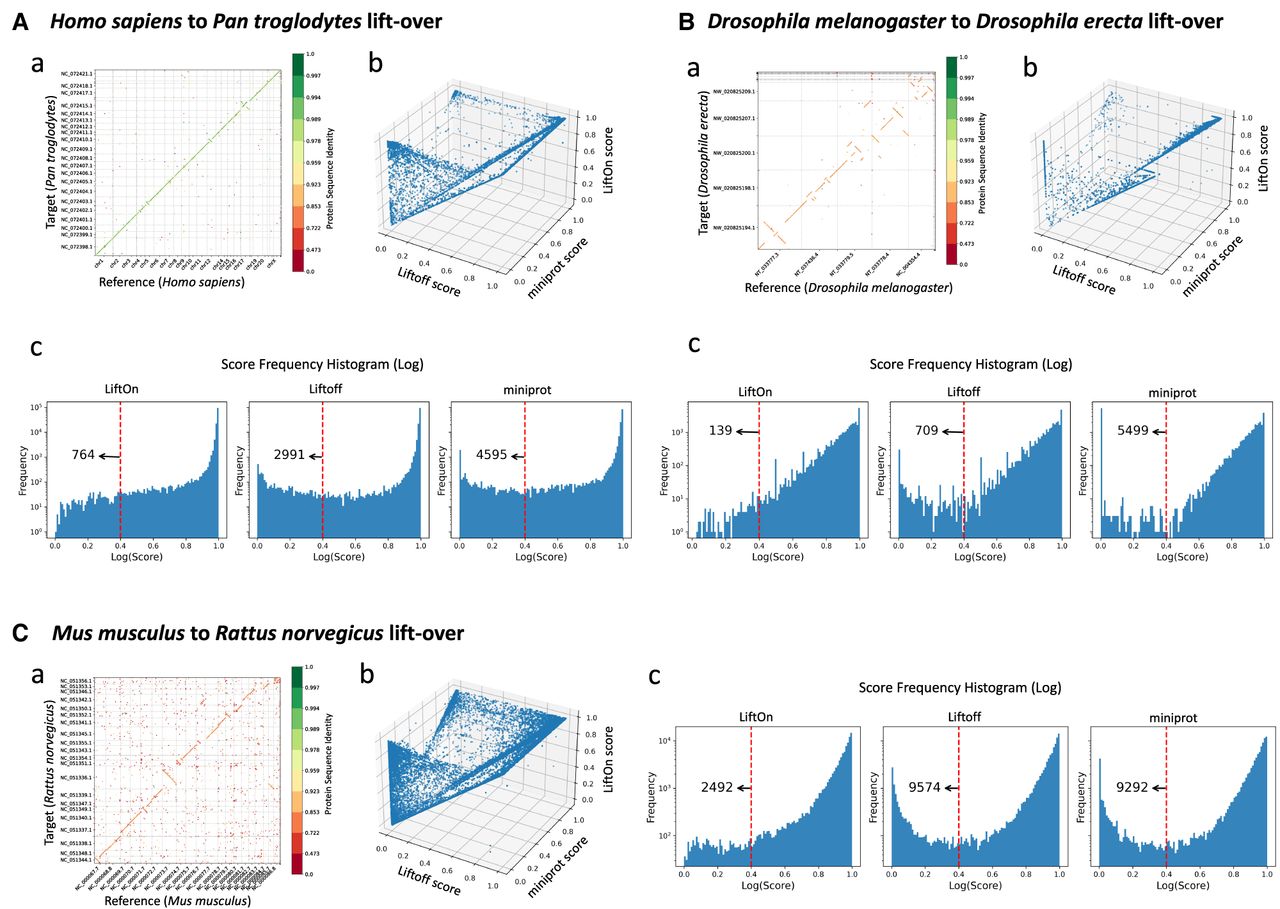
Chimpanzees are the closest evolutionary relatives of humans (Goodman 1999; Ebersberger et al. 2002; Chimpanzee Sequencing and Analysis Consortium 2005; Prüfer et al. 2012), so we chose this example to demonstrate the applicability of LiftOn between closely related species. LiftOn successfully lifted-over 37,509 genes (19,485 + 157 + 17,596 + 271) (Table 2), leaving 477 (285 + 192) genes unmapped, of which 285 were protein-coding and 192 noncoding. The overall gene mapping rate was 98.7%. Out of all mapped genes, 37,081 (19,485 + 17,596) were mapped uniquely, whereas 428 (157 + 271) genes were mapped to multiple locations, producing a total of 38,879 gene loci. LiftOn obtained higher protein sequence identity scores than Liftoff and miniprot for 4332 and 33,509 transcripts, respectively (Fig. 5A; Supplemental Note S7; Supplemental Fig. S13). We also compared LiftOn, Liftoff, and miniprot annotations to the published chimpanzee annotation, GCF_028858775.1_NHGRI_mPanTro3-v1.1-hic.freeze_pri, and found that LiftOn's results were superior to both Liftoff and miniprot as measured by matching intron chains and transcripts (Supplemental Table S11).
Evaluation of gene annotation lifting across species and tools using gene order plots, protein identity dot plots, and frequency distributions. (A) Comparative analysis of lifting over RefSeq v220 annotations from Homo sapiens (GRCh38) to Pan troglodytes (NHGRI_mPanTro3-v1.1). (B) Comparative analysis of lifting over annotations from Drosophila melanogaster (genome assembly release 6 + ISO1MT) to Drosophila erecta (Prin_Dsim_3.1). (C) Comparative analysis of lifting over annotations from Mus musculus (GRCm39) to Rattus norvegicus (mRatBN7.2). Graphs labeled a show protein-gene order plots, with the x-axis representing the reference genome and the y-axis representing the target genome. The protein sequence identities are color-coded on a logarithmic scale, ranging from green (one) to red (zero), and represent the degree of amino acid similarity, with one indicating identical sequences and zero indicating no shared amino acids. The gene order plot script was customized from LiftoffTools. Graphs labeled b are 3D protein sequence identity plots comparing Liftoff on the x-axis, miniprot on the y-axis, and LiftOn on the z-axis. Each dot represents a protein-coding transcript. If a dot is above the x = y plane, LiftOn's mapping produced a higher protein sequence identity score than the other programs. Graphs labeled c are frequency plots on a logarithmic scale of protein sequence identity for LiftOn (left), Liftoff (middle), and miniprot (right).
Statistics for LiftOn at both the gene and transcript levels, after mapping RefSeq release v220 annotation from the human genome to Pan troglodytes (mPanTro3-v1.1), from Drosophila melanogaster (genome assembly release 6 + ISO1MT) to Drosophila erecta (Prin_Dsim_3.1), and from Mus musculus (GRCm39) to Rattus norvegicus (mRatBN7.2)
| Lift-over experiment | Feature | Reference/Target | Total feature count | Protein-coding feature count | Noncoding feature count | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Single copy | Extra copy | Extra copy count | Total | Single copy | Extra copy | Extra copy count | Total | ||||
| Homo sapiens→Pan troglodytes | Gene | Reference (GRCh38) | 37,986 | 19,927 | 18,059 | ||||||
| Target (Pan_trog_v1) | 38,879 | 19,485 | 157 | 535 | 20,177 | 17,596 | 271 | 835 | 18,702 | ||
| Transcript | Reference (GRCh38) | 160,561 | 130,528 | 30,033 | |||||||
| Target (Pan_trog_v1) | 161,222 | 128,859 | 450 | 899 | 130,208 | 29,233 | 404 | 1377 | 31,014 | ||
| Drosophila melanogaster→Drosophila erecta | Gene | Reference (D. melanogaster) | 16,005 | 13,962 | 2043 | ||||||
| Target (D. erecta) | 15,276 | 13,180 | 140 | 152 | 13,472 | 1804 | 0 | 0 | 1804 | ||
| Transcript | Reference (D. melanogaster) | 33,176 | 30,749 | 2427 | |||||||
| Target (D. erecta) | 32,143 | 29,739 | 138 | 150 | 30,027 | 2116 | 0 | 0 | 2116 | ||
| Mus musculus→Rattus norvegicus | Gene | Reference (M. musculus) | 35,551 | 22,192 | 13,359 | ||||||
| Target (R. norvegicus) | 33,706 | 20,490 | 126 | 164 | 20,780 | 12,926 | 0 | 0 | 12,926 | ||
| Transcript | Reference (M. musculus) | 119,745 | 96,192 | 23,553 | |||||||
| Target (R. norvegicus) | 115,855 | 92,767 | 125 | 163 | 93,055 | 22,800 | 0 | 0 | 22,800 | ||
Mapping from D. melanogaster to D. erecta
We then mapped the annotation between two Drosophila species (Adams et al. 2000; Clark et al. 2007) that are considerably more distant from each other than human and chimpanzee—D. melanogaster (Hoskins et al. 2015) and D. erecta (Rio et al. 1983; David et al. 2007)—with 0.07 Dashing 2 (Baker and Langmead 2023) similarity score and 0.08 Mash-distance (Ondov et al. 2016). A protein-coding gene order plot (Fig. 5B) shows multiple genome rearrangements, illustrating the divergence between the genomes. The LiftOn gene- and transcript-level statistics are summarized in Table 2, showing a gene mapping rate of 94.5% and a transcript mapping rate of 96.4%. LiftOn yields improved annotations for 4583 and 7763 protein-coding transcripts compared with Liftoff and miniprot, respectively (Supplemental Note S8; Supplemental Fig. S14).
Mapping from M. musculus to R. norvegicus
Finally, we chose two model organisms, M. musculus (Church et al. 2011) and R. norvegicus (Howe et al. 2021) (mouse and rat), to showcase LiftOn's ability to map annotation between even more distant species, with 0.01 Dashing 2 (Baker and Langmead 2023) similarity score and 0.12 Mash-distance (Ondov et al. 2016). The results are illustrated in Figure 5C and Table 2, which reveal a gene mapping rate of 94.3% and a transcript mapping rate of 96.6%. For the mouse-to-rat mapping, LiftOn improves the annotations for 15,420 and 30,574 protein-coding transcripts compared with Liftoff and miniprot, respectively. The frequency plots in Figure 5C reveal that LiftOn only had 2492 protein-coding transcripts below the 40% identity threshold, a notable improvement over Liftoff (9574) and miniprot (9292). More details are included in Supplemental Note S9 and Supplemental Figures S15 and S16. We also compared LiftOn, Liftoff, and miniprot annotations to the published mRatBN7.2 rat annotation and found that LiftOn was superior in terms of matching intron chains and transcripts (Supplemental Table S12).
Discussion
Annotation lift-over is a powerful means to transfer annotation from one genome to another, but mapping requires accurate alignment, which increases in difficulty as genomes become more divergent. As the reference and target genomes become more distant, the improvement provided by LiftOn increases compared with Liftoff and miniprot approaches, as shown in Table 3. However, factors such as pseudogenes, large gene families, structural variations, and the quality of the source annotation all affect the difficulty of the problem. In terms of running times, LiftOn can transfer annotation between two human genomes in ∼23 min, and it can map human to chimpanzee in ∼4.5 h. Supplemental Table S13 provides a detailed breakdown of the run times for each major step.
The percentage of protein-coding transcripts mapped with varying degrees of protein sequence identity by LiftOn, Liftoff, and miniprot
| Experiment | LiftOn (%) (1/≥0.8/≥0.5) | Liftoff (%) (1/≥0.8/≥0.5) | miniprot (%) (1/≥0.8/≥0.5) |
|---|---|---|---|
| GRCh38.p14 to CHM13 | 64.4/84.3/84.6 | 64.3/84.2/84.3 | 49.7/82.5/83.4 |
| Human to Pan troglodytes | 17.8/83.5/84.6 | 17.8/82.5/83.2 | 13.4/80.7/82.0 |
| Mus musculus to Rattus norvegicus | 3.6/76.8/83.5 | 3.4/71.8/77.0 | 2.7/72.4/77.4 |
| Drosophila melanogaster to Drosophila erecta | 15.2/81.7/92.0 | 13.2/80.1/90.3 | 10.2/70.0/75.4 |
[i] Scores are categorized as 100% identity (score = 1), at least 80% identity (score ≥ 0.8), and at least 50% identity (score ≥ 0.5). The comparison evaluates the performance of the programs in mapping annotation from GRCh38.p14 to CHM13, human to P. troglodytes, M. musculus to R. norvegicus, and D. melanogaster to D. erecta.
LiftOn's accuracy will always be critically dependent on the accuracy of the source annotation. As an example, consider the mapping of two uncharacterized protein-coding genes, LOC124905331 and LOC112268317, both of which mapped onto CHM13 in multiple copies. LOC124905331 appears on an unplaced contig (NT_187388.1) in GRCh38, and on CHM13, LiftOn mapped it to Chromosome 21, spanning positions 3,205,883 to 5,522,421, with 50 tandemly repeated copies. LOC112268317 was initially on another unplaced contig (NT_187499.1) in GRCh38, and LiftOn mapped it in 30 copies on CHM13, near the telomeric regions of Chromosomes 14, 15, 21, and 22. Both of these genes overlap with the ribosomal DNA (rDNA) array, which occurs in many copies on the acrocentric Chromosomes 13, 14, 15, 21, and 22 (Agrawal and Ganley 2018; Nurk et al. 2022; Chao et al. 2023). Given this overlap with rDNA, we suspect that the source genes are not protein-coding genes, and manual curation would be required to clean up the results of the automated lift-over process.
LiftOn is a versatile homology-based annotation mapping tool, designed as a successor to Liftoff, the leading homology-based annotation lift-over tool. LiftOn improves on Liftoff by mapping protein-coding transcripts more accurately, which it achieves through the use of a protein-to-DNA aligner, miniprot; a chaining algorithm to generate protein sequences with high similarity to reference proteins; and postprocessing methods to identify and correct ORFs that might otherwise produce truncated proteins. Note that for noncoding genes, LiftOn uses the annotations generated by Liftoff. In its current implementation, LiftOn's chaining algorithm relies solely on miniprot alignments to refine Liftoff annotations. However, it could be extended in the future to take advantage of multiple DNA- or protein-based annotations or assembled RNA-seq transcripts. Although LiftOn currently relies on a single reference genome and its corresponding annotation, it could, in principle, use multiple source genomes.
Our experiments demonstrate that LiftOn outperforms other methods that rely solely on DNA or proteins for mapping annotations from one genome to another. Its use of protein sequence alignment allows it to effectively map annotation between different species, even those as distantly related as mouse and rat.
Methods
LiftOn is implemented as a Python package that maps gene features from a reference genome to a target genome using DNA-based and protein alignment–based methods, namely, Liftoff (Shumate and Salzberg 2021) and miniprot (Li 2023). For inputs, LiftOn requires a reference annotation in either GFF or GTF format, as well as reference and target genomes provided as FASTA files. LiftOn uses gffutils version 0.12 (https://github.com/daler/gffutils) to create a sqlite3 database, and it uses Biopython (Cock et al. 2009) and Pyfaidx (https://github.com/mdshw5/pyfaidx) to extract both DNA and protein transcript sequences from the reference genome. These extracted sequences, stored as intermediate FASTA files, serve as inputs for the embedded Liftoff code and miniprot module through a Python subprocess package. The outputs are separate Liftoff and miniprot annotations. Alternatively, users have the option to run Liftoff and miniprot separately and then input the annotations using the “‐‐liftoff” and “‐‐miniprot” arguments to LiftOn. The two annotations are then used to create gffutils sqlite3 databases.
Annotation lift-over special handling of the human reference
For all mappings of the human annotation, we excluded all alternative scaffolds and patches from the GRCh38 genome and its annotation. Specifically, we excluded scaffolds ending in “_fix” and “_alt,” because they are duplicates or variants of sequences found on the primary chromosomes. We also excluded rRNAs, which occur in hundreds of identical copies that vary widely among humans and create problems for the alignment programs (Agrawal and Ganley 2018; Hori et al. 2021; Chao et al. 2023).
Pairing Liftoff and miniprot protein-coding transcript annotations
Liftoff was designed to map any genomic interval, containing genes, transcripts, and exons, from one genome onto another. Liftoff's output uses a hierarchy in which genes contain one or more transcripts, and transcripts contain exons and/or CDS features. A CDS feature defines the CDSs within a protein-coding transcript; therefore, some CDS features span the same intervals as exons, whereas others span only parts of exons when the exons also include UTRs. In contrast, miniprot was designed to map protein sequences to a genome and can only generate annotations of transcripts containing CDS features.
After running both Liftoff and miniprot, LiftOn goes through the miniprot mappings and finds, for each one, the Liftoff genomic interval to which it corresponds. A miniprot transcript is considered to match a Liftoff transcript if (1) the loci of the two transcripts overlap, and (2) the locus of the miniprot transcript does not overlap with any other Liftoff gene loci, except those for which the matched Liftoff locus also overlaps. Note that the genomic intervals found by Liftoff are mostly distinct, but it does allow up to 10% overlap between adjacent gene features by default. On the other hand, miniprot lacks the capability to reconcile overlapping gene loci, which sometimes results in a protein-coding transcript from a large gene family mapping to multiple gene loci instead of one or to both a pseudogene and the correct locus.
In most cases, LiftOn is able to find a one-to-one mapping between the miniprot transcripts and those from LiftOff. For instance, mapping RefSeq release 220 annotations from GRCh38.p14 to T2T-CHM13 v2 results in 128,351 of 136,978 protein-coding transcripts (93.7%) being uniquely matched between Liftoff and miniprot (see Supplemental Note S10; Supplemental Fig. S17).
For the cases in which miniprot identifies multiple copies for a protein-coding transcript, LiftOn checks whether there is at least one copy overlapping with a Liftoff gene locus. Miniprot transcript mappings spanning multiple Liftoff loci are removed to prevent erroneous “read through” annotations. Among the remaining options, the transcript with the highest protein sequence identity score is selected. Note that in the GRCh38.p14 to T2T-CHM13 v2 lift-over there were 355 protein-coding transcripts in which none of the miniprot-discovered protein-coding transcripts overlapped with a corresponding Liftoff gene locus.
Once the one-to-one correspondence between two transcripts is established, LiftOn considers both Liftoff and miniprot CDS features of the two transcripts and initiates its PM algorithm as described in the next Methods section.
PM algorithm
The PM algorithm consists of two components: the chaining algorithm and the ORF search algorithm. The pseudocode for the protein_maximization function is outlined in Algorithm S1.
Step 1: the chaining algorithm
The chaining algorithm is a method for collecting and concatenating the optimal segments of CDSs obtained through the DNA-based mapping approach (Liftoff) and the protein-based approach (miniprot) into a consistent CDS chain, with the goal of maximizing the similarity of the mapped protein to the reference protein. In this context, the reference protein refers to the protein extracted from the reference annotation and genome, for example, the RefSeq annotation on GRCh38.
LiftOn uses Biopython (Cock et al. 2009) to extract and translate mapped protein-coding transcript sequences into their corresponding proteins and uses the Parasail (Daily 2016) Python package to align these protein sequences with the reference proteins. Following the alignment step, LiftOn maps the CDS boundaries onto the pairwise protein alignment results (see Fig. 1C). The algorithm initially maps the CDS boundaries onto the protein (get_cds_protein_boundary function in Algorithm S2) and then adjusts these boundaries using the cigar string obtained from the protein alignment if gaps exist in the target protein (adjust_cds_protein_boundary function in Algorithm S2).
Subsequently, the chaining algorithm clusters CDSs from Liftoff and miniprot. It starts by grouping the initial CDSs from both annotations until it encounters a boundary where the aligned amino acid counts in the reference protein up to that boundary match for both Liftoff and miniprot. These CDSs form a comparison block for which LiftOn calculates a partial protein sequence identity between the reference protein and those annotated by Liftoff and miniprot, as described in the Methods. CDS groups with higher identity scores relative to the reference protein are selected for the final LiftOn annotation. After this block is processed, LiftOn identifies a new group of CDSs starting from the previous found boundary and repeats the same selection process. The chaining algorithm's full pseudocode is provided in Algorithm S3.
Step 2: the ORF search algorithm
Following the chaining algorithm, LiftOn performs an ORF search algorithm on the protein-coding regions of the mapped transcripts that have mutations likely to be more deleterious, such as “frameshift,” “stop codon gain,” “stop codon loss,” and “start codon loss” mutations. The objective is to generate the longest valid protein sequences that align with the full-length reference proteins.
For each selected protein-coding transcript, LiftOn's ORF search algorithm iterates through three reading frames (zero, one, two). In each frame, it identifies potential ORFs by searching for start codons (“ATG”) and locating the corresponding stop codons (“TAA,” “TAG,” “TGA”) and retains the longest one it finds in that frame.
After identifying potential ORFs, LiftOn proceeds to compare the longest ORF in each frame to the reference protein sequence. It calculates and compares the sequence identity score of each candidate and selects the ORF with the highest sequence identity. If the selected ORF's identity exceeds the original annotation, LiftOn updates the CDS boundaries of the protein-coding transcript.
DNA and protein transcript sequence identity score calculation
To compute DNA sequence identity scores, LiftOn first extracts transcript sequences by concatenating all exonic regions in a transcript. Subsequently, it aligns each transcript sequence mapped on the target genome by LiftOn, Liftoff, or miniprot to its respective reference sequence. This alignment is performed using the nw_trace_scan_sat function from the Parasail (Daily 2016) Python package, configured with a match score of one, mismatch penalty of −3, gap opening penalty of two, and gap extension penalty of two. LiftOn then reports the percentage of identity between the two aligned sequences, defined as in BLAST (Altschul et al. 1990) as the number of matching bases in the two sequences over the number of alignment columns. The pseudocode of the algorithm described in this paragraph is illustrated in the get_DNA_id_fraction function from Algorithm S4. An example is provided in Supplemental Figure S18A.
To compute protein sequence identity scores, LiftOn first generates the protein sequence for each mapped transcript by translating the sequence obtained from concatenating all CDS regions in the transcript. Then, it aligns each protein sequence with the full-length reference protein by performing a pairwise alignment using the BLOSUM 62 matrix (Henikoff and Henikoff 1992), a gap opening penalty of 11 for insertions and deletions (indels), and a gap extension penalty of two. The protein sequence identity score is calculated up to the first encountered stop codon in the target protein. Differing slightly from the BLAST-style metric used for DNA sequence identity, LiftOn compresses the gaps in the reference alignment to prevent overpenalization of longer proteins in the target genome. These proteins might be mapped as longer because of potential repeat regions within the target genome or because of potential truncations in the proteins of the reference genome. The pseudocode for this process is presented in the get_AA_id_fraction function in Algorithm S4. An example is provided in Supplemental Figure S18B.
Additionally, the get_partial_id_fraction function in Algorithm S4 describes this process when the sequence identity computation is restricted to evaluating matches between partial proteins, as in the chaining algorithm, to determine the best matching CDS group with respect to the reference.
Resolving overlapping gene loci and finding extra copies of protein-coding genes
LiftOn uses both Liftoff and miniprot to identify additional copies of protein-coding genes, prioritizing Liftoff for its capability to also map UTR regions. First, LiftOn uses the embedded Liftoff module to iteratively map new gene copies and verify whether the lifted-over gene loci overlap with existing annotations. If so, it allows overlaps that are also present in the reference genome; if not, it permits a maximum overlap of 10% with other gene loci. The Liftoff module also ensures that each gene copy meets the user-specified minimum DNA sequence identity between the reference and target gene loci. This process continues until all possible copies have been identified.
Then, LiftOn applies the miniprot module to identify any additional copies of protein-coding genes that Liftoff might have missed. LiftOn uses the intervaltree package, a self-balancing interval tree implemented in Python (https://github.com/chaimleib/intervaltree), to manage gene loci intervals and enable fast detection of overlaps. Because miniprot maps annotations at the protein-coding transcript (mRNA) level, LiftOn copies the corresponding gene-level feature from the reference annotation as the parent of the mRNA feature identified exclusively by miniprot to ensure consistency within the “gene–mRNA–exon” hierarchy. LiftOn also enforces the constraint that any extra gene copies identified by miniprot should have at most a 10% overlap with other gene loci (computed as the ratio of the mapped miniprot length to the smaller of the two values: the mapped miniprot length or the reference protein length).
Furthermore, because miniprot maps only proteins and does not consider the UTRs in gene loci, it is very likely that miniprot maps to a processed pseudogene or only maps a partial gene segment. To remove potential pseudogenes, we implemented stricter filtering rules for gene loci identified exclusively by miniprot. First, if miniprot identifies only one CDS (i.e., with no intervening introns), the gene locus is annotated only if there is also a single CDS in the reference annotation. Second, the ratio of the coding regions in the miniprot annotation to the coding regions in the longest isoform of a reference gene locus must be between 0.9 and 1.5. Note that any extra gene copies identified exclusively by miniprot will not include UTRs.
LiftOn arguments used in the study
All analyses in this study were performed using LiftOn with its default parameters along with the additional arguments “-copies,” “-sc 0.95,” and “-polish.” The “-copies” argument prompts Liftoff to search for extra gene copies following the initial lift-over process. A gene copy will be annotated at a specific locus only if there is no overlap with an already annotated feature. Accompanying the “-copies” argument, the “-sc 0.95” parameter modifies the default Liftoff requirement of 100% sequence identity of mapped exons/CDSs to the reference ones, reducing it to allow a 95% sequence identity. The “-copies” argument will also trigger the annotation of extra copies of gene loci, which were distinctly found by miniport.
The “-polish” argument enables the Liftoff step in LiftOn to realign exons to correct CDSs that may have been altered during the lift-over process, such as the loss of start/stop codons or the introduction of in-frame stop codons, although this step extends the processing time.
To map RefSeq release v220 (results 2.2), MANE version 1.2 (Supplemental Note S1; Supplemental Table S4; Supplemental Fig. S7), and CHESS 3 (Supplemental Note S2; Supplemental Table S5; Supplemental Fig. S8) human annotations from GRCh38 to CHM13 (Table 1; Fig. 2), the “-chroms<chromosome_mapping.txt>” argument was also used in order to first perform a mapping step on a chromosome-by-chromosome basis. Once this step is complete, any genes that remain unmapped are remapped to the entire target assembly. This setting resulted in improved human annotation mapping accuracy. For all other experiments, LiftOn was run without the “-chroms” argument, mapping gene sequences to the full genome.
The “-f” argument and the input file “features.txt” indicate all types of parent features to lift over. To ensure LiftOn does not mistakenly identify pseudogenes as extra gene copies, we ran LiftOn with the “features.txt” file specifying both “gene” and “pseudogene” features to lift them over along with their children.
LiftOn runs miniprot with default parameters plus the “-gff-only” option to exclusively generate a GFF output file. Users can customize miniprot arguments to suit their specific data needs using the “-mp_options” parameter. This parameter should be followed by a string of options, separated by an “=” sign. The “=” is crucial, as it distinguishes miniprot options from Liftoff options that share the same flag. We recommend using the “-mp_options=“-j 2”” for splicing alignment in mammals and “-mp_options=“-j 1”” for nonmammals.
All experiments were conducted on a 24-core, 48-thread Intel Xeon Gold 6248R Linux computer with 1024 GB memory, using a single thread of execution.
Gene and transcript feature counting
The counts of protein-coding and noncoding genes and transcripts reported in this study were calculated as follows.
Gene counting
Gene features were classified as “protein-coding,” “noncoding,” and “others” based on the “gene_biotype” attribute in NCBI's RefSeq or the “gene_type” attribute in EMBL-EBI's Ensembl/GENCODE and CHESS 3. More specifically, a gene feature was categorized as a protein-coding gene if the feature type was “gene” and its type attribute was “protein_coding”; a gene was categorized as noncoding if its type attribute was either “lncRNA” or “ncRNA.” Other gene features, including “pseudogene,” “miRNA,” “snoRNA,” “tRNA,” “V_segment,” “snRNA,” “J_segment,” “misc_RNA,” “C_region,” and “antisense_RNA,” were categorized as “others.” The full list is provided in Supplemental Table S1.
Transcript counting
Transcript features were also categorized into “protein-coding,” “noncoding,” and “others.” The criteria for classification were based on both the type of transcript and the type of its parent gene. A transcript was counted as protein-coding if its feature type was “mRNA” and its parent gene was a protein-coding gene; a transcript was classified as noncoding if its feature type was either “lncRNA” or “ncRNA” and its parent gene was also a noncoding gene. The remaining transcripts were categorized as “others.”
Running LiftOn on a test data set
We provide sample files for the users wanting to test LiftOn at our GitHub repository: https://github.com/Kuanhao-Chao/LiftOn/tree/main/test. To execute LiftOn, the user can type a single command that uses the “-g” flag to specify a reference annotation file in GFF format (e.g., “NCBI_RefSeq_no_rRNA.gff”), the “-o” flag to specify the output file (e.g., “lifton.gff3”), plus both the reference genome file (e.g., “GCF_000001405.40_GRCh38.p14_genomic.fna”) and the target genome file (e.g., “chm13v2.0.fa”). For example, lifton -g NCBI_RefSeq_no_rRNA.gff -o lifton.gff3 -copies chm13v2.0.fa GCF_000001405.40_GRCh38.p14_genomic.fna.
Genomes used in this study
Brief rationales for selecting the genomes used in our experiments are described below. NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) accession numbers, assembly names, URLs for genome assembly and annotation downloads, and annotation release or last updated dates are summarized in Table 4.
Genome assembly and annotation details for species analyzed in this study
[i] Note that for each species, the table includes the GenBank accession number, assembly name, FTP sites for assembly and annotation downloads, and annotation release dates.
Apis mellifera
At the time of this study, GenBank contained no A. mellifera assemblies of the same strain as the reference genome, Amel_HAv3.1 (strain DH4) (Wallberg et al. 2019). We considered both ASM1932182v1 (subspecies ligustica) (Cao et al. 2021) and ASM1384124v2 (subspecies carnica) and chose ASM1932182v1 because it was more contiguous (42 contigs) than ASM1384124v2 (313 contigs).
Arabidopsis thaliana
The A. thaliana reference genome, TAIR10.1 (Lamesch et al. 2012), represents the Columbia (Col-0) strain. Of the other Col-0 assemblies, Col-CC and Col-CEN (ASM2311539v1) (Naish et al. 2021) were of similar quality, having all chromosomes completely assembled. We chose Col-CEN (ASM2311539v1) because Col-CC was a consensus assembly of 13 independent assemblies, whereas Col-CEN (ASM2311539v1) was derived from a single organism.
Mus musculus
At the time of this study, GenBank contained no high-quality contiguous assemblies of the same strain (C57BL/6J) as the reference genome, GRCm39 (Church et al. 2011). The only other assembly of the C57BL/6J strain (ASM377452v2) is quite fragmented, with 36,193 contigs. We chose NOD_SCID (strain NOD/SCID) (Schmid-Siegert et al. 2023) instead because it has the fewest contigs (281) of all other M. musculus assemblies from known strains.
Oryza sativa
The reference genome for rice, IRGSP-1.0 (Kawahara et al. 2013), belongs to the Japonica subgroup and the Nipponbare cultivar. From the three other assemblies that were also from the Nipponbare cultivar, we chose ASM3414082v1 (Shang et al. 2023) as it was the most contiguous, with every chromosome fully assembled into a single scaffold.
Software availability
LiftOn is implemented as a Python package. The LiftOn project is freely available on GitHub (https://github.com/Kuanhao-Chao/LiftOn) and on PyPi (https://pypi.org/project/LiftOn/).
The LiftOn source code can also be found as Supplemental Code. The LiftOn documentation is available at https://ccb.jhu.edu/lifton/.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all the members of the Salzberg and Pertea laboratories for their valuable discussions and insights. This research was supported in part by the U.S. National Institutes of Health under grants R01-HG006677 and R35-GM130151. J.M.H. is supported by the U.S. National Institutes of Health training grant T32HG002295.
Author contributions: K.-H.C., M.P., and S.L.S. designed the research. K.-H.C. and A.S. developed the LiftOn software. K.-H.C., J.M.H., A.M., M.P., and S.L.S. analyzed the results. K.-H.C. and J.M.H. conducted experiments on Liftoff and miniprot annotation mapping, involving both the same species and species that were closely and distantly related. K.-H.C. and C.H. visualized the results. K.-H.C. and A.M. wrote the LiftOn documentation. J.M.H. devised the name “LiftOn” for the software. A.M. designed the LiftOn logo. K.-H.C., J.M.H., C.H., M.P., and S.L.S. wrote the manuscript.
Notes
[4] Supplementary material [Supplemental material is available for this article.]
[5] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279620.124.
References
- ↵Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. 10.1126/science.287.5461.2185
- ↵Agrawal S, Ganley AR. 2018. The conservation landscape of the human ribosomal RNA gene repeats. PLoS One 13: e0207531. 10.1371/journal.pone.0207531
- ↵Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2
- ↵Baker DN, Langmead B. 2023. Genomic sketching with multiplicities and locality-sensitive hashing using Dashing 2. Genome Res 33: 1218–1227. 10.1101/gr.277655.123
- ↵Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. 2021. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3: lqaa108. 10.1093/nargab/lqaa108
- ↵Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, Lei J, Achawanantakun R, Jiao D, Lawrence CJ, 2014. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol 164: 513–524. 10.1104/pp.113.230144
- ↵Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18: 188–196. 10.1101/gr.6743907
- ↵Cao L, Zhao X, Chen Y, Sun C. 2021. Chromosome-scale genome assembly of the high royal jelly-producing honeybees. Sci Data 8: 302. 10.1038/s41597-021-01091-7
- ↵Chao K-H, Zimin AV, Pertea M, Salzberg SL. 2023. The first gapless, reference-quality, fully annotated genome from a southern Han Chinese individual. G3 (Bethesda) 13: jkac321. 10.1093/g3journal/jkac321
- ↵Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. 10.1038/nature04072
- ↵Chinwalla AT, Cook LL, Delehaunty KD, Fewell GA, Fulton LA, Fulton RS, Members of the Mouse Genome Analysis G. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. 10.1038/nature01262
- ↵Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, Chen H-C, Agarwala R, McLaren WM, Ritchie GR, 2011. Modernizing reference genome assemblies. PLoS Biol 9: e1001091. 10.1371/journal.pbio.1001091
- ↵Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218. 10.1038/nature06341
- ↵Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423. 10.1093/bioinformatics/btp163
- ↵Daily J. 2016. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinformatics 17: 81. 10.1186/s12859-016-0930-z
- ↵Danchin A, Ouzounis C, Tokuyasu T, Zucker JD. 2018. No wisdom in the crowd: genome annotation in the era of big data—current status and future prospects. Microb Biotechnol 11: 588–605. 10.1111/1751-7915.13284
- ↵David JR, Lemeunier F, Tsacas L, Yassin A. 2007. The historical discovery of the nine species in the Drosophila melanogaster species subgroup. Genetics 177: 1969–1973. 10.1534/genetics.104.84756
- ↵Ebersberger I, Metzler D, Schwarz C, Pääbo S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet 70: 1490–1497. 10.1086/340787
- ↵Fiddes IT, Armstrong J, Diekhans M, Nachtweide S, Kronenberg ZN, Underwood JG, Gordon D, Earl D, Keane T, Eichler EE, 2018. Comparative annotation toolkit (CAT)—simultaneous clade and personal genome annotation. Genome Res 28: 1029–1038. 10.1101/gr.233460.117
- ↵Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, 2019. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47: D766–D773. 10.1093/nar/gky955
- ↵Goodman M. 1999. The genomic record of humankind's evolutionary roots. Am J Hum Genet 64: 31–39. 10.1086/302218
- ↵Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci 89: 10915–10919. 10.1073/pnas.89.22.10915
- ↵Hoff KJ, Lomsadze A, Borodovsky M, Stanke M. 2019. Whole-genome annotation with BRAKER. Methods Mol Biol 1962: 65–95. 10.1007/978-1-4939-9173-0_5
- ↵Hori Y, Shimamoto A, Kobayashi T. 2021. The human ribosomal DNA array is composed of highly homogenized tandem clusters. Genome Res 31: 1971–1982. 10.1101/gr.275838.121
- ↵Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, 2015. The release 6 reference sequence of the Drosophila melanogaster genome. Genome Res 25: 445–458. 10.1101/gr.185579.114
- ↵Howe K, Dwinell M, Shimoyama M, Corton C, Betteridge E, Dove A, Quail MA, Smith M, Saba L, Williams RW, 2021. The genome sequence of the Norway rat, Rattus norvegicus Berkenhout 1769. Wellcome Open Research 6: 118. 10.12688/wellcomeopenres.16854.1
- ↵Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, 2013. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6: 4. 10.1186/1939-8433-6-4
- ↵Kovaka S, Ou S, Jenike KM, Schatz MC. 2023. Approaching complete genomes, transcriptomes and epi-omes with accurate long-read sequencing. Nat Methods 20: 12–16. 10.1038/s41592-022-01716-8
- ↵Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109
- ↵Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, 2012. The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40: D1202–D1210. 10.1093/nar/gkr1090
- ↵Li H. 2023. Protein-to-genome alignment with miniprot. Bioinformatics 39: btad014. 10.1093/bioinformatics/btad014
- ↵Logsdon GA, Vollger MR, Eichler EE. 2020. Long-read human genome sequencing and its applications. Nat Rev Genet 21: 597–614. 10.1038/s41576-020-0236-x
- ↵Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, 2013. The Genotype-Tissue Expression (GTEx) Project. Nat Genet 45: 580–585. 10.1038/ng.2653
- ↵Marx V. 2023. Method of the year: long-read sequencing. Nat Methods 20: 6–11. 10.1038/s41592-022-01730-w
- ↵Morales J, Pujar S, Loveland JE, Astashyn A, Bennett R, Berry A, Cox E, Davidson C, Ermolaeva O, Farrell CM, 2022. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604: 310–315. 10.1038/s41586-022-04558-8
- ↵Naish M, Alonge M, Wlodzimierz P, Tock AJ, Abramson BW, Schmücker A, Mandáková T, Jamge B, Lambing C, Kuo P, 2021. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374: eabi7489. 10.1126/science.abi7489
- ↵Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987
- ↵O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189
- ↵Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. 2016. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17: 132. 10.1186/s13059-016-0997-x
- ↵Prüfer K, Munch K, Hellmann I, Akagi K, Miller JR, Walenz B, Koren S, Sutton G, Kodira C, Winer R, 2012. The bonobo genome compared with the chimpanzee and human genomes. Nature 486: 527–531. 10.1038/nature11128
- ↵Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35(Database issue): D61–D65. 10.1093/nar/gkl842
- ↵Rhie A, Nurk S, Cechova M, Hoyt SJ, Taylor DJ, Altemose N, Hook PW, Koren S, Rautiainen M, Alexandrov IA, 2023. The complete sequence of a human Y chromosome. Nature 621: 344–354. 10.1038/s41586-023-06457-y
- ↵Rio B, Couturier G, Lemeunier F, Lachaise D. 1983. Evolution d'une spécialisation saisonniére chez Drosophila erecta (Dipt., Drosophilidae). Ann Soc Ent Fr (NS) 19: 235–248. 10.1080/21686351.1983.12278361
- ↵Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754
- ↵Salzberg SL. 2019. Next-generation genome annotation: we still struggle to get it right. Genome Biol 20: 92. 10.1186/s13059-019-1715-2
- ↵Schmid-Siegert E, Qin M, Tian H, Arpat B, Chen B, Xenarios I. 2023. Reference genomes for BALB/c nude and NOD/SCID mouse models. G3 (Bethesda) 13: jkad188. 10.1093/g3journal/jkad188
- ↵Shang L, He W, Wang T, Yang Y, Xu Q, Zhao X, Yang L, Zhang H, Li X, Lv Y, 2023. A complete assembly of the rice Nipponbare reference genome. Mol Plant 16: 1232–1236. 10.1016/j.molp.2023.08.003
- ↵Sharma V, Schwede P, Hiller M. 2017. CESAR 2.0 substantially improves speed and accuracy of comparative gene annotation. Bioinformatics 33: 3985–3987. 10.1093/bioinformatics/btx527
- ↵Shendure J, Balasubramanian S, Church GM, Gilbert W, Rogers J, Schloss JA, Waterston RH. 2017. DNA sequencing at 40: past, present and future. Nature 550: 345–353. 10.1038/nature24286
- ↵Shumate A, Salzberg SL. 2021. Liftoff: accurate mapping of gene annotations. Bioinformatics 37: 1639–1643. 10.1093/bioinformatics/btaa1016
- ↵Shumate A, Salzberg S. 2022. LiftoffTools: a toolkit for comparing gene annotations mapped between genome assemblies. F1000Res 11: 1230. 10.12688/f1000research.124059.1
- ↵Shumate A, Zimin AV, Sherman RM, Puiu D, Wagner JM, Olson ND, Pertea M, Salit ML, Zook JM, Salzberg SL. 2020. Assembly and annotation of an Ashkenazi human reference genome. Genome Biol 21: 129. 10.1186/s13059-020-02047-7
- ↵Stein L. 2001. Genome annotation: from sequence to biology. Nat Rev Genet 2: 493–503. 10.1038/35080529
- ↵Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics 14: 178–192. 10.1093/bib/bbs017
- ↵Van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. 2018. The third revolution in sequencing technology. Trends Genet 34: 666–681. 10.1016/j.tig.2018.05.008
- ↵Varabyou A, Sommer MJ, Erdogdu B, Shinder I, Minkin I, Chao K-H, Park S, Heinz J, Pockrandt C, Shumate A, 2023. CHESS 3: an improved, comprehensive catalog of human genes and transcripts based on large-scale expression data, phylogenetic analysis, and protein structure. Genome Biol 24: 249. 10.1186/s13059-023-03088-4
- ↵Wallberg A, Bunikis I, Pettersson OV, Mosbech M-B, Childers AK, Evans JD, Mikheyev AS, Robertson HM, Robinson GE, Webster MT. 2019. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics 20: 275. 10.1186/s12864-019-5642-0
- ↵Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat Genet 45: 1113–1120. 10.1038/ng.2764
- ↵Yandell M, Ence D. 2012. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet 13: 329–342. 10.1038/nrg3174
- ↵Zimin AV, Shumate A, Shinder I, Heinz J, Puiu D, Pertea M, Salzberg SL. 2022. A reference-quality, fully annotated genome from a Puerto Rican individual. Genetics 220: iyab227. 10.1093/genetics/iyab227