
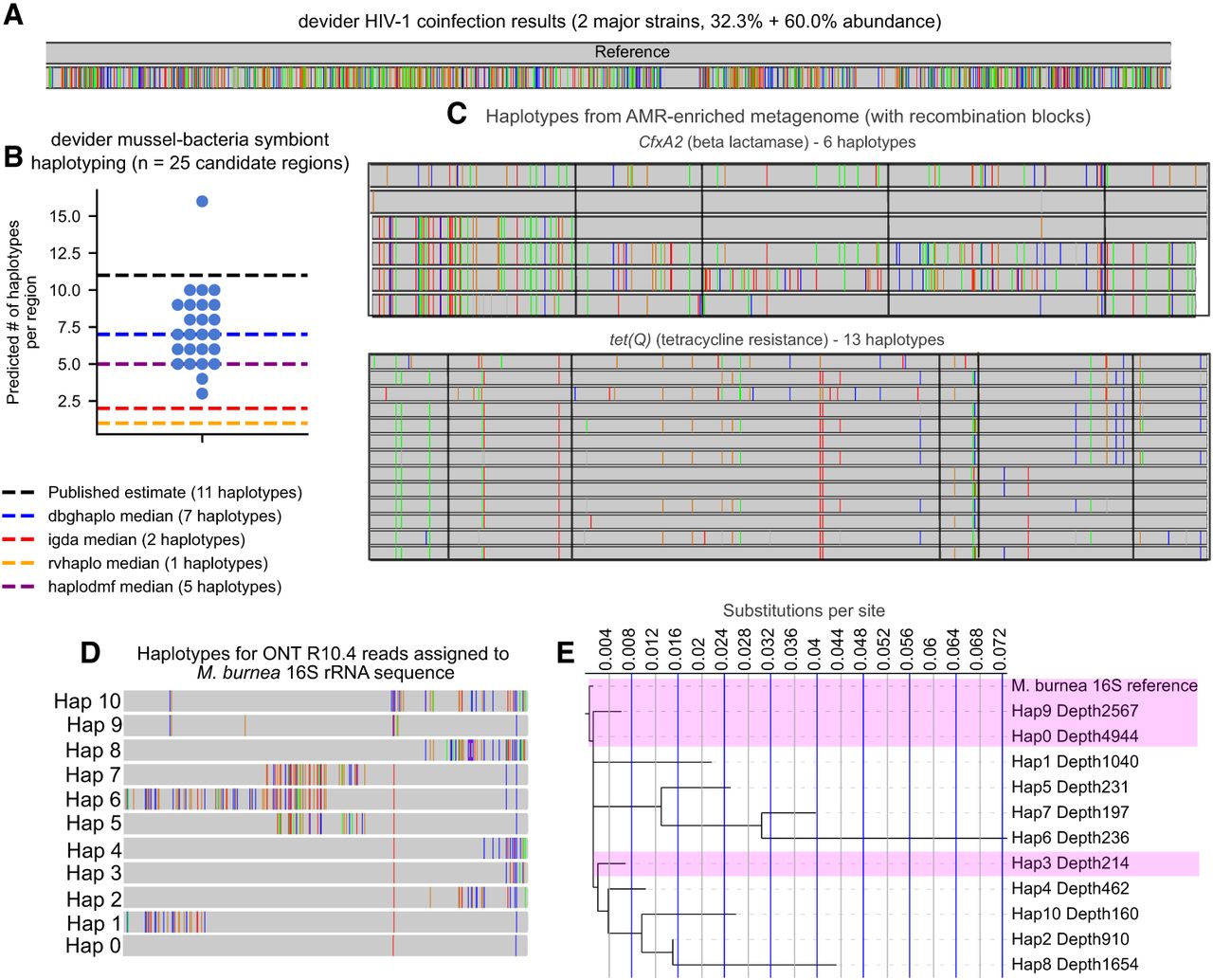
Long-read haplotyping results from real samples subjected to a variety of sequencing technologies. (A) Haplotypes from long-read HIV-1 nanopore sequencing (93%–94.2% predicted sequencing accuracy) of an HIV coinfection from Mori et al. (2022). Two major haplotypes were found by devider, confirming previous results. Mismatched bases are shown with the reference as the upper haplotype. (B) Haplotyping results for PacBio RS II sequencing (89.5% mean gap-compressed identity against reference) of an intracellular bacterial symbiont community within deep-sea mussels from Ansorge et al. (2019), who predicted 11 strains to be present. Twenty-five candidate single-copy regions with high SNP diversity were haplotyped by devider, iGDA, and RVHaplo; CliqueSNV was excluded because it timed out on multiple regions. devider produced higher diversity estimates compared with iGDA and RVHaplo, which both produced three or fewer haplotypes across all sites. (C) devider haplotyping of a long-read bovine gut metagenome enriched for AMR genes. CfxA2 (3200× coverage) and tet(Q) (19,500× coverage and last 1000 bp shown) haplotype sequences with >30× coverage and 1% abundance are shown with mismatches against their reference sequences in MEGARes v3.0. Mismatches shared by all haplotypes are removed. Recombination blocks are outlined in black as predicted by GARD. (D) devider haplotypes from an ONT R10.4 16S rRNA data set for the reference 16S sequence of the most abundant species Massilia burnea. (E) Phylogenetic tree of haplotypes assigned to the M. burnea reference. Depth of coverage is shown next to the haplotype ID. The x-axis shows the branch length from the root. Highlighted haplotypes have >99% identity to the reference.








