
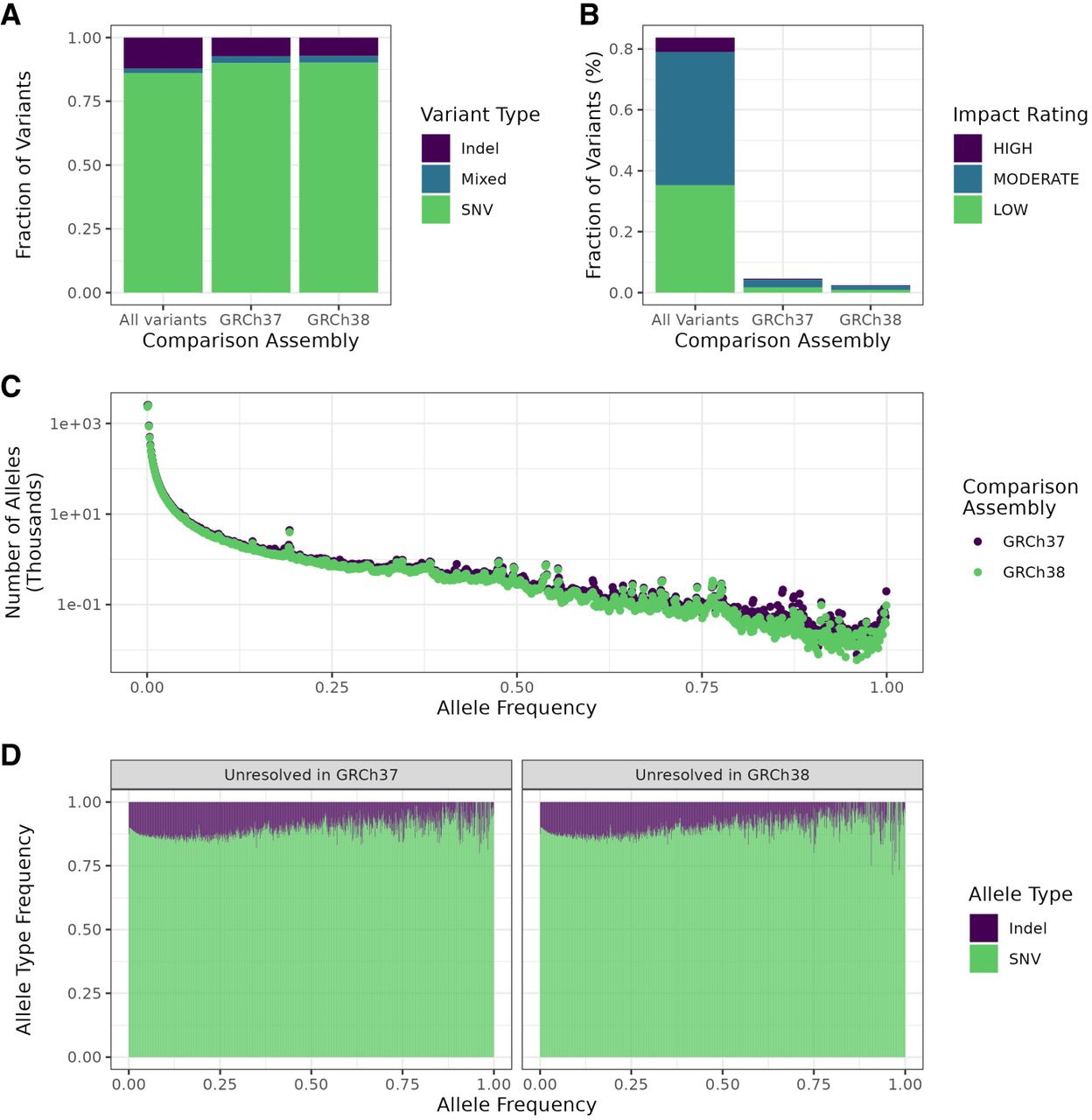
Summaries of variant characteristics from the whole cohort, restricted to newly resolved regions. (A) Fraction of variants by type in the novel regions in the T2T-CHM13 assembly that were unresolved in the reference as shown on the x-axis and the whole genome. (B) Fractions of variants by predicted impact in the whole call set as determined by SnpEff in previously unresolved regions and the whole genome. Variants with impact rating MODIFIER were excluded. (C) Distribution of allele frequencies for T2T-CHM13 specific variants mapped to regions not resolved in the two previous assemblies. (D) Fraction of each variant type by allele frequency. Frequencies were rounded to the closest multiple of 0.001 (1/1000).








